Циклоизомеризация - Cycloisomerization
Циклоизомеризация есть ли изомеризация в которой циклический изомер субстрата производится в координате реакции. Самым большим преимуществом реакций циклоизомеризации является их атомная экономичность, по замыслу, ничего не теряется, так как каждый атом исходного материала присутствует в продукте. В большинстве случаев эти реакции опосредованы катализатор на основе переходного металла, в некоторых случаях органокатализаторы и редко они возникают в тепловых условиях. Эти циклизации могут быть выполнены с превосходным уровнем селективности во многих случаях и превратили циклоизомеризацию в мощный инструмент для уникального и сложного молекулярного строительства.[1] Циклоизомеризация - очень широкая тема в органическом синтезе, и существует множество реакций, которые можно отнести к таковым. Два основных класса этих реакций: внутримолекулярный Майкл дополнение и внутримолекулярный Дильс – Альдер реакции. В рамках циклоизомеризации реакции циклоизомеризации енинов и родственных олефинов являются наиболее широко используемыми и изученными реакциями.[2]
Внутримолекулярное добавление Михаэля
Довольно интуитивно понятный путь к циклическим изомерам - внутримолекулярный сопряженное сложение к α, β – ненасыщенным карбонилы (внутримолекулярное добавление Майкла или IMA). Компетентные акцепторы Михаэля включают конъюгированные еноны, енали или производные нитроалкена, а примеры других акцепторов немногочисленны.[3] Несмотря на то, что реакции IMA в синтезе повсеместны, существует очень мало примеров асимметричных превращений IMA.[2]
Тиомочевина Катализаторы с боковыми хиральными цепями, как было показано, активируют системы с привязанными нитроалкановыми и сложноэфирными мотивами, вызывая асимметричный IMA.[3][4] Полезность этой трансформации была продемонстрирована при синтезе циклических предшественников γ– аминокислот (рис. 1).[3] Предполагается, что активация происходит через водородную связь нитроната и сложного эфира с тиомочевинным катализатором, что объясняет интересную селективность E–Эстер.[3]

Функциональный стереодивергентный органокатализируемый IMA /лактонизация трансформация в синтезе замещенных дигидрофуранов и тетрагидрофуранов была изучена на предмет ее способности создавать важные структурные мотивы во многих природных продуктах (рис. 2).[5] Когда эфиры, такие как 3 подчиняются (S) - (-) - гидрохлориду тетрамизола (4) катализатор результат син–2,3-замещенный ТГФ, в то время как дополнительный анти–Продукт легко доступен через алкалоидный катализатор хинного дерева, такой как 7.[5]

Внутримолекулярный метод Дильса-Альдера
Связанные пары внутримолекулярных реакций Дильса – Альдера (IMDA) диены и диенофилы в виде [4 + 2], наиболее распространенным из которых является терминальное замещение. Эти превращения популярны в тотальном синтезе и уже получили широкое распространение для множества сложных синтетических мишеней.[6] Одним из таких применений является применение энантиоселективной трансформации IMDA в асимметричном синтезе морского токсина (-) - изопуло’упона (10).[7]

Синтез (-) - изопуло’упона продемонстрировал полезность реакций IMDA, катализируемых катионным комплексом Cu (II) бис (оксазолин), для получения бициклических продуктов с четырьмя соседними стереогенными центрами (рис. 3).[7] Довольно недавнее применение IMDA-реакций в синтезе сложных молекул - подход IMDA к трициклическому ядру алкалоидов палинина ликоподия, класса натуральных продуктов, выделенных из клубного мха.[8]
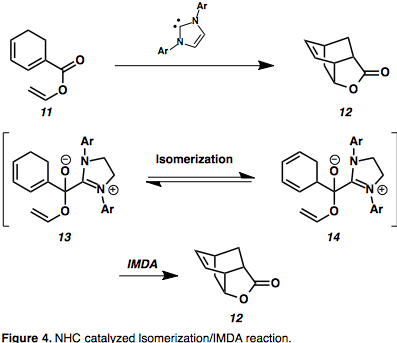
N – гетероциклические карбены (NHC) представляют собой новый класс органокатализаторов, способных индуцировать Umpolung реакционная способность, а также нормальные преобразования полярности, однако до недавнего времени они не широко использовались в полном синтезе из-за ограниченного объема субстрата.[9] Интересным расширением использования этих органокатализаторов является катализируемая NHC изомеризация олефинов / каскадная реакция IMDA с образованием уникальных бициклических каркасов.[10][11] Диениловые эфиры, такие как 11 были превращены в замещенные бицикло [2.2.2] октаны с помощью стадии изомеризации, стабилизированной полуацеталь-азолиевым промежуточным соединением (13).[11] Активационный барьер изомеризации 1,3-гексадиена посредством [1,5] -двига составляет 41 ккал / моль и, как ожидается, будет увеличиваться при конъюгации со сложным эфиром, поэтому некаталитическая изомеризация маловероятна.[11] Это дает преимущество обхода высокого барьера активации, обеспечивая доступ к ранее недоступным производным IMDA.
Циклоизомеризация энна
Циклоизомеризация енинов, алкиновый вариант реакции Альдерена (рис. 5), представляет собой внутримолекулярную перегруппировку 1, n-енинов с образованием соответствующего циклического изомера.
Хотя перегруппировка может происходить в термических условиях, объем термической перегруппировки ограничен из-за требований высоких температур, поэтому переходные металлы, такие как Au, Pd, Pt, Rh и Ir, часто используются в качестве катализаторов.[2] Поскольку синтез ссорится, чтобы построить сложные структурные мотивы в присутствии индуктивных, стереоэлектронных и стерических требований, эта перегруппировка недавно была разработана как надежный метод конструирования карбо- и гетероциклических каркасов с превосходными химио-, регио- и диастереоселективными результатами.[2]
Не существует единого механизма, который можно было бы использовать для описания циклоизомеризации енинов, поскольку механизм зависит от условий реакции и выбора катализатора.[2][12] Возможны промежуточные соединения циклоизомеризации, катализируемой металлом, в которой металл координирует алкин или алкен, активируя один или оба, и показаны на фиг.6.
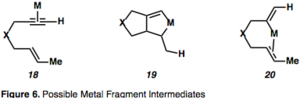
Активация алкина комплексообразованием с металлом, приводящая к η2–Металлический промежуточный продукт, такой как 18 открывает алкин для нуклеофильной атаки и порождает карбокатион промежуточные звенья. Эта Pull-push реактивность важна для понимания реакций, опосредованных π-кислотами. Комплексообразование алкина с металлическим фрагментом истощает электронную плотность в связи («притягивание»), в сочетании со способностью металла возвращаться назад («толкать») вызывает наблюдаемые последовательные электрофильный и нуклеофильный характер вицинальных атомов углерода алкин (рисунок 7).
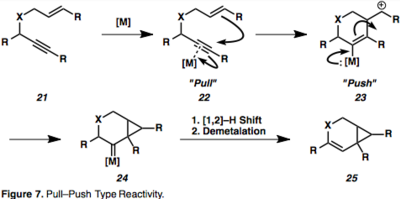
Промежуточные продукты металлацикла (19) являются результатом одновременного комплексообразования и активации обоих партнеров. Также возможно гидрометаллирование алкина с образованием соединений винилового металла, которые могут, в свою очередь, карбометаллировать олефин (20). Пример циклоизомеризации 1,6-енина, протекающей через η2–Активированный промежуточный металл представлен на рисунке 8,[13] что является обычным для циклоизомеризаций енинов, опосредованных Pt или Au, из-за их π-кислотной природы. Примечательно, что в этом примере имеет место перенос хиральности, при котором абсолютная стереохимия енина (26) контролирует стереохимию продукта (27).[13]
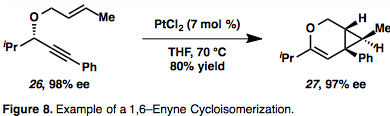
Циклоизомеризация енинов, опосредованная Au и Pt
Активация алкинов π-кислотными металлами, такими как Au или Pt, является традиционным методом в комплексном синтезе органических соединений, однако то, как эта активация влияет на реакционную способность, полностью не изучено, и, таким образом, механизм в значительной степени предлагается на основе результатов реакции и теоретических расчетов.[14][15] Катионные катализаторы Au (I) и Pt (II) являются привлекательным выбором, поскольку они демонстрируют сильные Кислота Льюиса характер и способность стабилизировать катионные промежуточные соединения, будучи стабильными.[16]
Универсальной функцией циклоизомеризации енинов, катализируемой Au (I), является создание асимметричных колец среднего размера, что является проблемой при синтезе орнаментированной молекулярной конструкции. Удобный доступ к асимметричным 7- и 8-членным карбоциклам возможен с использованием хирального золотого катализатора BINAP Au (I), дающего широкий спектр продуктов.[17]
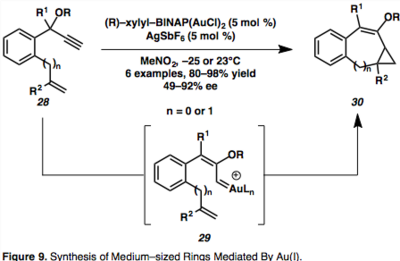
Предполагается, что внутримолекулярное циклопропанирование происходит через 1,2-сдвиг пропаргилового эфира, опосредованного Au, с образованием син–Au винилкарбеноиды (29).[17] Вычислительные исследования показывают, что син-средний, 29, образуется под кинетическим контролем, и предполагается, что он находится в равновесии с термодинамически благоприятным СНГ- промежуточное соединение, которое может быть перехвачено нуклеофилом, приводя к образованию винилциклопропандиеновых продуктов, однако это выходит за рамки данной статьи.[17]
Винилциклоалкены - еще один функциональный класс продуктов, доступных путем алкиновой активации енинов π-кислотными металлами. PtCl2 было показано, что он катализирует образование различных экзотических винилциклоалкенов из легкодоступных исходных материалов (рис. 10).[18]

Примечательно, что расширение цикла наблюдается для енинов с циклическими алкеновыми мотивами. Это объясняется формальным введением метиленовой группы олефина между двумя атомами углерода алкина; также было предложено механистическое объяснение этого расширения кольца.[19] Образование этих виниклоалкенов в сочетании с его способностью к расширению кольца было использовано для создания промежуточных 36 en путь к стрепторубину B.[18] Подобное превращение возможно с использованием катионных комплексов Au (I), однако здесь можно выбрать продукты винициклоалкена с помощью механизма, проходящего через начальную 5 – exo – dig или велосипедпропаны могут производиться с помощью начального 6 – эндо – копать.[20] Посредством вычислений ДПФ предполагается, что 5 – exo – dig циклизация благоприятна для комплексов Au (I), так как она имеет более низкий активационный барьер по сравнению с 6 – эндо – копать и действительно многочисленные примеры продуктов винилциклоалкенов, произведенных 5 – exo – dig приведены (рисунок 11).

Реакционная способность может быть изменена путем тщательного выбора условий реакции, выбора катализатора и субстрата.[20] Различная реакционная способность этих циклосиомеризаций, катализируемых переходными металлами, дополнительно демонстрирует их синтетическую полезность для построения уникальных молекулярных скелетов.
Рекомендации
- ^ Обзор реакций циклоизомеризации см .: Энантиоселективная циклоизомеризация, катализируемая переходными металлами Анджела Маринетти, Элен Жюльен и Арно Войтуриес Chem. Soc. Ред., 2012,41, 4884-4908 Дои:10.1039 / C2CS35020C, Критический обзор .
- ^ а б c d е Watson, I. D. G .; Toste, F. D. Chem. Sci. 2012, 3, 2899–2919.
- ^ а б c d Узлы, W. J .; Nutt, D. R .; Chippindale, A.M .; Cobb, A. J. A. J. Am. Chem. Soc. 2009, 131, 16016–16017.
- ^ Обзор катализа тиомочевины см .: Zhang, Z .; Schreiner, P.R. Chem. Soc. Ред. 2009, 38, 1187–1198.
- ^ а б Belmessieri, D .; Houpliere, A .; Calder, E. D. D .; Тейлор, Дж. Э .; Smith, A. D. Chem. Евро. J. 2014, 20, 9762–9769.
- ^ Такао, К; Munakata, R .; Tadano, K. Chem. Ред. 2005, 105, 4779–4807.
- ^ а б Johnson, J. S .; Evan, D. A. J. Org. Chem. 1997, 62, 786–787.
- ^ Сайзмор, N .; Рыхновский, С. Д. Орг. Lett. 2014, 16, 688–691.
- ^ Искьердо, Дж. Хатсон, Г. Э .; Коэн, Д. Т .; Scheidt, K. Angew. Chem. Int. Эд. 2012, 51, 11686–11698.
- ^ Для обзора каскадных реакций органокатализа N – гетероциклического карбена см .: Grossman, A .; Эндерс, Д. Энджью. Chem. Int. Эд. 2012, 51, 314–325.
- ^ а б c Ковальчик, М .; Lupton, D. W. Angew. Chem. Int. Эд. 2014, 53, 5314–5317.
- ^ Genêt, J. –P .; Toullec, P. Y .; Мишле, В. Ангью. Chem. Int. Эд. 2008, 47, 4268–4315.
- ^ а б Newcomb, E.T .; Ferreira, E. M. Org. Lett. 2013, 15, 1772–1775.
- ^ Fürstner, A .; Дэвис, П. У. Энджью. Chem. Int. Эд. 2007, 46, 3410–3449.
- ^ Обзор реакций циклоизомеризации енинов, катализируемых золотом, см .: Jiménez-Núñez, E .; Echavarren, A.M. Chem. Ред. 2008, 108, 3326–3350.
- ^ Горин, Д. Дж .; Тосте, Ф. Д. Природа 2007, 446, 395–403.
- ^ а б c Watson, I. D. G .; Риттер, S; Toste, F. D. J. Am. Chem. Soc. 2009, 131, 2056–2057.
- ^ а б Fürstner, A .; Stelzer, F .; Szillat, H. J. Am. Chem. Soc. 2001, 123, 11863–11869.
- ^ Zhang, L .; Sun, J .; Козьмин, С.А. Adv. Synth. Катал. 2006, 348, 2271–2296.
- ^ а б Nieto-Oberhuber, C .; Muñoz, M. P .; Buñel, E .; Nevado, C .; Cμrdenas, D. J .; Эчаваррен, А. М. Энгью. Chem. Int. Эд. 2004, 43, 2402–2406.