Перестановка Пейна - Payne rearrangement
В Перестановка Пейна это изомеризация, в основных условиях из 2,3-эпоксидных спиртов в изомерные 2,3-эпоксидные спирты с обращением конфигурации. Также известны аза- и тиа-пейновские перегруппировки азиридинов и тиираниумов соответственно.[1]
Вступление
В основных, протонных условиях 2,3-эпоксидные спирты претерпевают перегруппировку, в которой кислород спирта открывает эпоксид с изменением конфигурации, образуя изомерный 1,2-эпоксидный спирт. В целом перегруппировка Пейна представляет собой миграцию эпоксида. Хотя сама миграция полностью обратима, нуклеофильное раскрытие в условиях Куртина-Хэммета обеспечивает хорошие выходы функционализированных диолов, полученных из одного изомера эпоксидного спирта.[2] Внутримолекулярное электрофильное улавливание нового алкоксида, образующегося при перегруппировке, также может быть использовано для доведения реакции до завершения. В некоторых случаях термодинамическая разница между изомерами эпоксидов достаточно велика, чтобы получить один изомер с синтетически полезным выходом, не полагаясь на кинетические различия, связанные с захватом.
(1)

Для достижения уравновешивания требуются строго базовые условия, что ограничивает синтетическую применимость трансформации субстратами, не имеющими лабильной по основанию функциональности. Многие эпоксидные спирты равновесия очень хорошо сбалансированы;[3] однако использование описанных выше стратегий захвата может привести к высоким выходам единичных изомеров.
Механизм и стереохимия
Преобладающий механизм
Основной механизм перегруппировки Пейна включает депротонирование свободной гидроксильной группы, обратную нуклеофильную атаку на проксимальный углерод эпоксида и повторное протонирование только что освобожденного алкоксида. Каждый шаг процесса обратим.[4]
(2)

Некоторые наблюдения предполагают, что эта механистическая картина чрезмерно упрощена. Миграция эпоксида либо не происходит, либо очень медленная в апротонных условиях.[3]- было высказано предположение, что нуклеофильная атака замедляется за счет координации ионов металлов с нуклеофильным кислородом в апротонных условиях. Кроме того, когда внешний нуклеофил добавляется к уравновешивающим изомерам эпоксида, соотношение открытых продуктов не отражает соотношение изомеров эпоксида в растворе или их относительную термодинамическую стабильность.[5] На месте нуклеофильное раскрытие уравновешивающихся эпоксидов является примером Условия Куртина-Хэммета - поскольку эпоксиды быстро уравновешиваются относительно скорости раскрытия эпоксида, это кинетические барьеры раскрытия кольца которые контролируют наблюдаемое соотношение продукта. В приведенном ниже примере продукт раскрытия концевого эпоксида является основным продуктом, хотя сам концевой эпоксид менее термодинамически стабилен, чем внутренний изомер.
(3)

Галогендиолы можно использовать в качестве предшественников 2,3-эпоксидных спиртов перед перегруппировкой. Проблемы селективности сайта могут возникнуть, если две гидроксильные группы, фланкирующие галогенид, не эквивалентны. В общем, образование внутренних замещенных эпоксидов происходит быстрее, чем образование концевых эпоксидов.[6] Эта идея может быть использована для прогнозирования хода миграции на месте-генерированные эпоксиды.
(4)

Стереохимия
Перегруппировка Пейна происходит с инверсией стереохимии в C-2. Субстраты, содержащие несколько соседних гидроксильных групп, могут подвергаться «каскадной» миграции эпоксидов с инверсией в каждом месте нуклеофильной атаки. В одном примере инверсия трех смежных стереоцентров возникает после двух миграций эпоксида, раскрытия эпоксида карбоксилатом и гидролиза полученного лактона.[7]
(5)

Объем и ограничения
Перестановка Пейна
Положение равновесия как в циклических, так и в ациклических системах может быть предсказано по структурам двух уравновешивающих эпоксидов. В ациклических системах установлены такие правила:[8]
- Предпочтительно большее замещение в эпоксидном кольце.
- Среди дизамещенных эпоксидов транс изомеры предпочтительнее СНГ изомеры.
- Предпочтительны изомеры с первичными гидроксильными группами.
- Электронодонорные заместители на эпоксиде являются стабилизирующими, а электроноакцепторные заместители дестабилизируют.
Пиранозиды - наиболее изученные циклические системы. Исследования миграции эпоксидов в пиранозидах и других циклических эпоксидных спиртах выявили три обобщения:
- Как и в ациклических системах, более предпочтительным является замещение эпоксидного кольца.
- Предпочтительный изомер - это изомер с большим количеством псевдоэкваториальных заместителей.
- Внутримолекулярные водородные связи и другие взаимодействия в пространстве не играют роли в равновесных соотношениях.
Конформационно заблокированные пиранозиды показывают термодинамическое предпочтение циклических субстратов более псевдоэкваториальным группам.[9]
(6)

В апротонных условиях нуклеофильное раскрытие изомеров эпоксида может быть достигнуто с помощью гидридов или органокупратов. Нуклеофильная атака обычно происходит по меньшей мере по замещенному углероду, давая более замещенный диольный продукт.[10]
(7)

В протических условиях также обычно приветствуется открытие наименее замещенной позиции. Нуклеофилы, которые можно использовать в протонных условиях, включают фенолы, вторичные амины, азид-анион и сульфиды.[11]
(8)

Межмолекулярный нуклеофильный захват одного изомера эпоксида затруднен, поскольку реакция эпоксидного спирта с электрофилом обычно происходит быстрее, чем миграция. Тем не мение, внутримолекулярная электрофия часто эффективна для захвата одного изомера эпоксида. Например, второй соседний эпоксид в исходном материале уравнения (9) захватывается одним изомером эпоксида, что приводит к тетрагидрофуран.[12]
(9)

Аза- и тиа-пейн перегруппировки
Аза-пейновская перегруппировка может осуществляться либо в «прямом» (эпоксид в азиридин), либо в «обратном» (азиридин в эпоксид) направлении в зависимости от используемых условий. Бедные электронами азиридины претерпевают обратную перегруппировку в присутствии гидридного основания,[13] в то время как соответствующие эпоксиамины претерпевают прямую перегруппировку в присутствии эфирата трифторида бора.[14]
(10)

Перегруппировка тиа-Пейна наблюдалась только в прямом направлении (эпоксид в тииран) с на месте открытие тиирана. Обратное нуклеофильное раскрытие на C-2 возможно за счет использования реагентов триалкилалюминия.[15]
(11)

Синтетические приложения
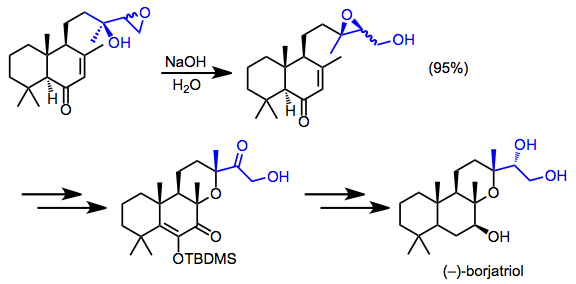
Синтез боржатриола включал редкое выделение мигрировавшего эпоксида. Смесь диастереомеров продуктов перегруппировки использовали до конца синтеза.[16]
(12)
Последние две стадии полного синтеза шпатола включали внутримолекулярное электрофильное улавливание алкоксида, полученного из перегруппированного эпоксида. Атака промежуточного алкоксида на соседний мезилат давала бис (эпоксид), а дебензилирование давало целевое соединение.[17]
(13)

Сравнение с другими методами
Другие методы, доступные для получения 2,3-эпоксидных спиртов, имеют то преимущество, что они не начинаются с существующего 2,3-эпоксидного спирта; однако они, как правило, включают больше стадий, чем миграция эпоксида. Асимметричное дигидроксилирование можно использовать для синтеза эпоксидных спиртов с высокой стереоселективностью, а некоторые методы, основанные на дигидроксилировании, избегают использования сильно основных условий.[18]
(14)

Альтернативный метод, который приводит к сохранению конфигурации на C-2, включает мезилирование эпоксидного спирта, открытие эпоксида и повторное закрытие путем замещения мезилата.[11]
(15)

Условия и порядок экспериментов
Типичные условия
В условиях перегруппировки может происходить раскрытие концевых эпоксидов случайным гидроксидом; если это нежелательно, следует использовать безводные растворители, реагенты и стеклянную посуду. Свежеприготовленный метоксид натрия в метаноле обычно используется для осуществления перегруппировки без раскрытия. Нуклеофильное раскрытие может быть достигнуто за счет использования азид натрия, избыток гидроксида или купратных реагентов в присутствии хлорид лития. Электрофильный захват осуществляется в стандартных условиях в присутствии электрофила, такого как бензилбромид. Силилгалогениды также использовались в качестве электрофильных улавливающих агентов.
Чтобы предотвратить миграцию эпоксида, можно использовать слабоосновные условия. Ни водный карбонат калия, ни водные аминовые основания не вызывают перегруппировку эпоксида. Низкие температуры также полезны, когда миграция эпоксида нежелательна.
Пример процедуры[19]
(16)

Раствор метил (циано) купрата (раствор A) готовили следующим образом: к суспензии 0,35 г (3,91 ммоль) цианида меди (I) в 5 мл тетрагидрофурана в атмосфере аргона при 0 ° по каплям в течение примерно 5 минут добавляли. 2,76 мл раствора метиллития в этиловом эфире (1,4 М, 3,86 ммоль). Бесцветный раствор перемешивали в течение 10 минут при 0 °, нагревали до 25 ° в течение 30 минут, затем снова охлаждали до 0 °. Отдельно готовили раствор литиевой соли (±) -цис-4-бензилокси-2,3-эпокси-1-бутанола (раствор Б) следующим образом: к раствору 0,5 г (2,58 ммоль) эпоксидной смолы. спирт и 0,90 г (21,4 ммоль) хлорида лития в 10 мл тетрагидрофурана в атмосфере аргона при -78 ° по каплям добавляли 1,65 мл раствора н-бутиллития в гексане (1,56 M, 2,58 ммоль). Раствор перемешивали в течение 5 минут при -78 °, давали нагреться до 0 °, а затем перемешивали при этой температуре в течение 10 минут. Реакцию осуществляли добавлением раствора A к раствору B через канюлю при 0 ° с последующим нагреванием до комнатной температуры в течение 2 часов. Затем реакционную смесь перемешивали еще 12 часов и затем осторожно обрабатывали 5 мл насыщенного водного раствора. хлорид аммония. Смесь перемешивали в течение 1-2 часов для облегчения удаления остатков меди. Затем добавляли этиловый эфир (20 мл) и отделяли органический слой. Водную фазу дважды экстрагировали 20 мл этилового эфира, и объединенные органические фазы сушили над сульфат магния, фильтровали и концентрировали с получением 0,51 г продукта в виде бесцветного масла (95%), ИК (пленка) 3400, 3100, 3060, 3030, 2970, 2930, 2870, 1600, 1500, 1465, 1445, 1385, 1370 , 1320, 1285, 1210, 1180, 1120, 1100, 1075, 1030, 1020, 980, 905, 830, 750, 730, 710, 695 см – 1; 1H ЯМР (CDCl3) δ 0,90 (т, J = 6,0 Гц, 3 H), 1,37–1,53 (м, 2 H), 3,20 (ш с, 2 H), 3,40–3,65 (м, 4 H), 4,48 (с, 2 H) ), 7,29 (с, 5 H).
Рекомендации
- ^ Хэнсон, Р. Орг. Реагировать. 2002, 60, 1. Дои:10.1002 / 0471264180.or060.01
- ^ Симан, Дж. И. Chem. Ред. 1983, 83, 83.
- ^ а б Пейн, Г. Б. J. Org. Chem. 1962, 27, 3819.
- ^ Angyal, S.J .; Гилхэм, П. Т. J. Chem. Soc. 1957, 3691.
- ^ Кацуки, Т .; Ли, А. В. М .; Карта.; Мартин, В. С .; Masamune, S .; Шарплесс, К. Б .; Tuddenham, D .; Уокер, Ф. Дж. J. Org. Chem. 1982, 47, 1373.
- ^ Paulsen, H .; Эберштейн, К. Chem. Бер. 1976, 109, 3891.
- ^ Bock, K .; Lundt, I .; Педерсен, К. Carbohydr. Res. 1988, 179, 87.
- ^ Pierre, J.-L .; Chautemps, P .; Арно, П. Бык. Soc. Чим. Пт. 1969, 106, 1317.
- ^ Мубарак, А .; Фрейзер-Рид, Б. J. Org. Chem. 1982, 47, 4265.
- ^ Page, P. C. B .; Rayner, C.M .; Сазерленд, И. О. J. Chem. Soc., Perkin Trans. 1 1990, 1375.
- ^ а б Behrens, C.H .; Ko, S. Y .; Шарплесс, К. Б .; Уокер, Ф. Дж. J. Org. Chem. 1985, 50, 5687.
- ^ Klein, E .; Rojahn, W .; Хеннеберг, Д. Тетраэдр 1964, 20, 2025.
- ^ Harden, R.C .; Hodgkinson, T. J .; McKillop, A .; Prowse, W. G .; Уркхарт, М. В. Дж. Тетраэдр 1997, 53, 21.
- ^ Nakai, K .; Ибука, Т .; Отака, А .; Tamamura, H .; Fujii, N .; Ямамото, Ю. Tetrahedron Lett. 1995, 36, 6247.
- ^ Сасаки, М .; Танино, К .; Мияшита, М. J. Org. Chem. 2001, 66, 5388.
- ^ Herlem, D .; Хуонгхуу, Ф. Тетраэдр 1997, 53, 673.
- ^ Soloman, R.G .; Basu, B .; Рой, С .; Сачинуала, Н. Д. Варенье. Chem. Soc. 1991, 113, 3096.
- ^ Ko, S. Y .; Малик, М. Tetrahedron Lett. 1993, 34, 4675.
- ^ Page, P. C. B .; Rayner, C.M .; Сазерленд, И. О. J. Chem. Soc., Perkin Trans. 1 1990, 1375.