Виртуальный просмотр - Virtual screening
Виртуальный просмотр (ПРОТИВ) - вычислительный метод, используемый в открытие лекарств искать библиотеки маленькие молекулы чтобы определить те структуры, которые с наибольшей вероятностью будут связываться с мишень для наркотиков обычно белок рецептор или же фермент.[2][3]
Виртуальный скрининг определяется как «автоматическая оценка очень больших библиотек соединений» с использованием компьютерных программ.[4] Как следует из этого определения, VS в основном представляла собой числовую игру, в которой основное внимание уделялось тому, как огромные химическое пространство из более 1060 мыслимые соединения[5] можно отфильтровать до управляемого числа, которое можно синтезировать, купить и протестировать. Хотя поиск по всей химической вселенной может быть теоретически интересной проблемой, более практичные сценарии VS сосредоточены на разработке и оптимизации целевых комбинаторных библиотек и обогащении библиотек доступных соединений из собственных репозиториев соединений или предложений поставщиков. По мере увеличения точности метода виртуальный скрининг стал неотъемлемой частью открытие лекарств процесс.[6][1] Виртуальный скрининг можно использовать для выбора соединений из внутренней базы данных для скрининга, выбора соединений, которые могут быть приобретены за пределами США, и выбора соединения, которое следует синтезировать следующим.
Методы
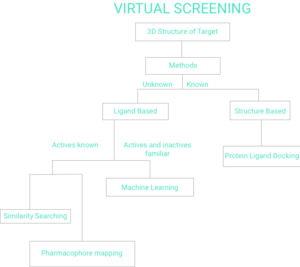
Существует две широких категории методов скрининга: на основе лиганда и на основе структуры.[7] Остальная часть этой страницы будет отражать блок-схему виртуального скрининга на Рисунке 1.
На основе лиганда
Учитывая набор структурно разнообразных лиганды что привязано к рецептор, модель рецептора может быть построена путем использования коллективной информации, содержащейся в таком наборе лигандов. Они известны как фармакофор модели. Затем кандидатный лиганд можно сравнить с моделью фармакофора, чтобы определить, совместим ли он с ним и, следовательно, может ли он связываться.[8]
Другой подход к виртуальному скринингу на основе лигандов заключается в использовании 2D методов анализа химического сходства.[9] для сканирования базы данных молекул на предмет наличия одной или нескольких активных лигандных структур.
Популярный подход к виртуальному скринингу на основе лигандов основан на поиске молекул, форма которых аналогична форме известных активных веществ, поскольку такие молекулы будут соответствовать сайту связывания мишени и, следовательно, будут связываться с мишенью. В литературе имеется ряд перспективных приложений этого класса методов.[10][11][12] Фармакофорные расширения этих 3D-методов также бесплатно доступны в виде веб-серверов.[13][14]
На основе структуры
Виртуальный скрининг на основе структуры включает стыковка кандидатов-лигандов в белок-мишень с последующим применением функция подсчета очков чтобы оценить вероятность того, что лиганд свяжется с белком с высоким сродством.[15][16][17] Веб-серверы, ориентированные на перспективный виртуальный просмотр, доступны всем.[18][19]
Гибридные методы
Гибридные методы, основанные на сходстве структур и лигандов, также были разработаны для преодоления ограничений традиционных подходов VLS. В этой методологии используется эволюционная информация о связывании лигандов для прогнозирования низкомолекулярных связывающих веществ.[20][21] и может использовать как глобальное структурное сходство, так и карманное подобие.[20] Подход, основанный на глобальном структурном сходстве, использует как экспериментальную структуру, так и предсказанную модель белка, чтобы найти структурное сходство с белками в библиотеке голотемплат PDB. После обнаружения значительного структурного сходства, метрика коэффициента Танимото на основе двумерных отпечатков пальцев применяется для скрининга небольших молекул, которые похожи на лиганды, извлеченные из выбранных шаблонов holo PDB.[22][23] Предсказания этого метода были экспериментально оценены и показывают хорошее обогащение в идентификации активных малых молекул.
Указанный выше метод зависит от глобального структурного подобия и не способен априори выбор конкретного сайта связывания лиганда в интересующем белке. Кроме того, поскольку методы основаны на оценке двухмерного сходства лигандов, они не способны распознавать стереохимическое сходство небольших молекул, которые существенно различаются, но демонстрируют сходство геометрической формы. Чтобы решить эти проблемы, новый карманный подход, PoLi, способные к нацеливанию на специфические связывающие карманы в голо-белковых матрицах, были разработаны и экспериментально оценены.
Вычислительная инфраструктура
Вычисление парных взаимодействий между атомами, которое является предпосылкой для работы многих программ виртуального скрининга, необходимо вычислительная сложность, где N - количество атомов в системе. Из-за квадратичного масштабирования по отношению к количеству атомов вычислительная инфраструктура может варьироваться от портативного компьютера для метода на основе лиганда до мэйнфрейма для метода на основе структуры.

На основе лиганда
Методы на основе лигандов обычно требуют доли секунды для одной операции сравнения структур. Одного процессора достаточно, чтобы выполнить большой просмотр за несколько часов. Однако можно провести несколько сравнений параллельно, чтобы ускорить обработку большой базы данных соединений.
На основе структуры
Размер задачи требует параллельные вычисления инфраструктура, например, кластер Linux системы, запускающие процессор пакетной очереди для обработки работы, например Sun Grid Engine или крутящий момент PBS.
Требуются средства обработки ввода из больших составных библиотек. Для этого требуется форма составной базы данных, которая может запрашиваться параллельным кластером, доставляя соединения параллельно различным вычислительным узлам. Коммерческие механизмы баз данных могут быть слишком громоздкими, а высокоскоростной механизм индексирования, например Berkeley DB, может быть лучшим выбором. Кроме того, может быть неэффективным запускать одно сравнение для каждого задания, потому что время разгона узлов кластера может легко превысить объем полезной работы. Чтобы обойти это, необходимо обрабатывать пакеты соединений в каждом задании кластера, объединяя результаты в своего рода файл журнала. Вторичный процесс для анализа файлов журналов и извлечения кандидатов с высокими показателями может быть запущен после того, как весь эксперимент будет проведен.
Точность
Цель виртуального скрининга - выявить молекулы новой химической структуры, которые связываются с макромолекулярными молекулами. цель интереса. Таким образом, успех виртуального экрана определяется с точки зрения поиска новых интересных каркасов, а не общего количества совпадений. Поэтому к интерпретациям точности виртуального скрининга следует относиться с осторожностью. Низкий процент попаданий интересных каркасов явно предпочтительнее, чем высокие показатели успешности уже известных каркасов.
Большинство тестов виртуальных скрининговых исследований в литературе ретроспективны. В этих исследованиях эффективность метода VS измеряется его способностью извлекать небольшой набор ранее известных молекул, обладающих сродством к интересующей мишени (активные молекулы или просто активные компоненты), из библиотеки, содержащей гораздо более высокую долю предполагаемых неактивных или приманки. Напротив, в перспективных приложениях виртуального скрининга полученные совпадения подвергаются экспериментальному подтверждению (например, IC50 измерения). Существует консенсус в отношении того, что ретроспективные эталоны не являются хорошими предикторами предполагаемой эффективности, и, следовательно, только проспективные исследования представляют собой убедительное доказательство пригодности метода для конкретной цели.[24][25][26][27][28]
Приложение к открытию лекарств
Виртуальный скрининг - очень полезное приложение, когда дело доходит до идентификации молекул-ударов как основы медицинской химии. По мере того как подход виртуального скрининга становится все более важным и существенным методом в индустрии медицинской химии, этот подход быстро расширяется.[29]
Методы на основе лигандов
Не зная структуры, пытаюсь предсказать, как лиганды будут связываться с рецептором. С использованием фармакофорных особенностей каждый лиганд идентифицируется как донор, так и акцепторы. Приравнивающие элементы накладываются друг на друга, однако маловероятно, что существует единственное правильное решение.[1]
Фармакофорные модели
Этот метод используется при объединении результатов поиска с использованием разных эталонных соединений, одинаковых дескрипторов и коэффициентов, но разных активных соединений. Этот метод полезен, потому что он более эффективен, чем использование одной эталонной структуры вместе с наиболее точной производительностью, когда дело касается различных активных объектов.[1]
Фармакофор - это совокупность стерических и электронных свойств, которые необходимы для оптимального супрамолекулярного взаимодействия или взаимодействий с биологической структурой-мишенью, чтобы вызвать ее биологический ответ. Выберите представителя как набор активных, большинство методов будут искать похожие привязки. Желательно иметь несколько жестких молекул, и лиганды должны быть разнообразными, другими словами, обеспечить наличие различных свойств, которые не проявляются во время фазы связывания.[1]
Структура
Постройте составную прогнозную модель на основе известных активных и известных неактивных знаний. QSAR (количественно-структурная взаимосвязь деятельности), которая ограничена небольшим однородным набором данных. SAR (отношение структуры и активности), где данные обрабатываются качественно и могут использоваться со структурными классами и более чем одним режимом привязки. Модели отдают предпочтение соединениям для обнаружения потенциальных клиентов.[1]
Машинное обучение
Чтобы использовать машинное обучение для этой модели виртуального скрининга, необходим обучающий набор с известными активными и известными неактивными соединениями. Также существует модель деятельности, которая затем вычисляется посредством субструктурного анализа, рекурсивного разбиения, опорных векторных машин, k ближайших соседей и нейронных сетей. Последний шаг - определение вероятности того, что соединение является активным, а затем ранжирование каждого соединения на основе его вероятности быть активным.[1]
Субструктурный анализ в машинном обучении
Первая модель машинного обучения, используемая для больших наборов данных, - это анализ субструктуры, который был создан в 1973 году. Каждая субструктура фрагмента вносит непрерывный вклад в действие определенного типа.[1] Субструктура - это метод, который преодолевает трудности, связанные с огромной размерностью, когда дело доходит до анализа структур при разработке лекарств. Эффективный анализ каркаса используется для конструкций, которые имеют сходство с многоуровневым зданием или башней. Геометрия используется для нумерации граничных швов данной конструкции в начале и ближе к кульминации. При разработке метода специальной статической конденсации и процедур замещения этот метод оказался более продуктивным, чем предыдущие модели анализа субструктур.[30]
Рекурсивное разбиение
Рекурсивное разделение - это метод, который создает дерево решений с использованием качественных данных. Понимание того, как правила разбивают классы с небольшой ошибкой неправильной классификации при повторении каждого шага до тех пор, пока не будет найдено разумных разделений. Однако рекурсивное разбиение может иметь плохую предсказательную способность, потенциально создавая точные модели с той же скоростью.[1]
Структурные методы стыковки известных белковых лигандов
Лиганд может связываться с активным сайтом в белке, используя алгоритм поиска стыковки и функцию подсчета очков, чтобы идентифицировать наиболее вероятную причину для отдельного лиганда при назначении порядка приоритета.[1][31]
Смотрите также
- Грид-вычисления
- Скрининг с высокой пропускной способностью
- Док (молекулярный)
- Функции подсчета очков
- База данных ZINC
Рекомендации
- ^ а б c d е ж грамм час я j Жилле V (2013). «Виртуальный скрининг на основе лигандов и структур» (PDF). Университет Шеффилда.
- ^ Rester U (июль 2008 г.). «От виртуальности к реальности - виртуальный скрининг при обнаружении и оптимизации потенциальных клиентов: перспектива медицинской химии». Текущее мнение в области открытия и разработки лекарств. 11 (4): 559–68. PMID 18600572.
- ^ Роллингер Дж. М., Ступпнер Х, Лангер Т. (2008). «Виртуальный скрининг для открытия биологически активных натуральных продуктов». Природные соединения как лекарства Том I. Прогресс в исследованиях лекарств. Fortschritte der Arzneimittelforschung. Progres des Recherches Pharmaceutiques. Прогресс в исследованиях лекарств. 65. С. 211, 213–49. Дои:10.1007/978-3-7643-8117-2_6. ISBN 978-3-7643-8098-4. ЧВК 7124045. PMID 18084917.
- ^ Уолтерс В.П., Шталь М.Т., Мурко М.А. (1998). «Виртуальный просмотр - обзор». Drug Discov. Сегодня. 3 (4): 160–178. Дои:10.1016 / S1359-6446 (97) 01163-X.
- ^ Бохачек Р.С., Макмартин С., Гуида В.К. (1996). «Искусство и практика структурного дизайна лекарств: перспектива молекулярного моделирования». Med. Res. Rev. 16 (1): 3–50. Дои:10.1002 / (SICI) 1098-1128 (199601) 16: 1 <3 :: AID-MED1> 3.0.CO; 2-6. PMID 8788213.
- ^ МакГрегор MJ, Ло З, Цзян X (11 июня 2007 г.). «Глава 3: Виртуальный скрининг при открытии лекарств». В Хуан Z (ред.). Исследование открытия наркотиков. Новые рубежи в постгеномную эпоху. Wiley-VCH: Вайнхайм, Германия. С. 63–88. ISBN 978-0-471-67200-5.
- ^ Макиннес К. (октябрь 2007 г.). «Стратегии виртуального скрининга в открытии лекарств». Современное мнение в области химической биологии. 11 (5): 494–502. Дои:10.1016 / j.cbpa.2007.08.033. PMID 17936059.
- ^ Вс Х (2008). «Виртуальный скрининг на основе фармакофоров». Современная лекарственная химия. 15 (10): 1018–24. Дои:10.2174/092986708784049630. PMID 18393859.
- ^ Виллет П., Барнард Дж. М., Даунс Г. М. (1998). «Поиск химического сходства». Журнал химической информации и компьютерных наук. 38 (6): 983–996. CiteSeerX 10.1.1.453.1788. Дои:10.1021 / ci9800211.
- ^ Раш Т.С., Грант Дж. А., Мосяк Л., Николлс А. (март 2005 г.). «Основанный на форме 3-мерный метод перескока каркаса и его применение к бактериальному межбелковому взаимодействию». Журнал медицинской химии. 48 (5): 1489–95. CiteSeerX 10.1.1.455.4728. Дои:10.1021 / jm040163o. PMID 15743191.
- ^ Баллестер PJ, Westwood I, Laurieri N, Sim E, Richards WG (февраль 2010 г.). «Перспективный виртуальный скрининг со сверхбыстрым распознаванием формы: идентификация новых ингибиторов ариламин-N-ацетилтрансфераз». Журнал Королевского общества, Интерфейс. 7 (43): 335–42. Дои:10.1098 / rsif.2009.0170. ЧВК 2842611. PMID 19586957.
- ^ Кумар А., Чжан К.Ю. (2018). «Достижения в разработке методов подобия форм и их применение в открытии лекарств». Границы химии. 6: 315. Bibcode:2018Пт .... 6..315K. Дои:10.3389 / fchem.2018.00315. ЧВК 6068280. PMID 30090808.
- ^ Ли Х, Люнг К.С., Вонг М.Х., Баллестер П.Дж. (июль 2016 г.). «USR-VS: веб-сервер для крупномасштабного перспективного виртуального скрининга с использованием сверхбыстрых методов распознавания форм». Исследования нуклеиновых кислот. 44 (W1): W436–41. Дои:10.1093 / нар / gkw320. ЧВК 4987897. PMID 27106057.
- ^ Сперандио О., Петижан М., Туффери П. (июль 2009 г.). "wwLigCSRre: сервер на основе трехмерных лигандов для идентификации и оптимизации попаданий". Исследования нуклеиновых кислот. 37 (Выпуск веб-сервера): W504–9. Дои:10.1093 / nar / gkp324. ЧВК 2703967. PMID 19429687.
- ^ Kroemer RT (август 2007 г.). «Дизайн лекарств на основе структуры: стыковка и оценка». Современная наука о белках и пептидах. 8 (4): 312–28. CiteSeerX 10.1.1.225.959. Дои:10.2174/138920307781369382. PMID 17696866.
- ^ Кавасотто К.Н., Орри А.Дж. (2007). «Стыковка лигандов и виртуальный скрининг на основе структуры в открытии лекарств». Актуальные темы медицинской химии. 7 (10): 1006–14. Дои:10.2174/156802607780906753. PMID 17508934.
- ^ Kooistra AJ, Vischer HF, McNaught-Flores D, Leurs R, de Esch IJ, de Graaf C (2016). «Функционально-специфический виртуальный скрининг лигандов GPCR с использованием комбинированного метода оценки». Научные отчеты. 6: 28288. Bibcode:2016НатСР ... 628288K. Дои:10.1038 / srep28288. ЧВК 4919634. PMID 27339552.
- ^ Ирвин Дж. Дж., Шойчет Б. К., Мизингер М. М., Хуанг Н., Колицци Ф., Вассам П., Цао И (сентябрь 2009 г.). «Автоматическая стыковка экранов: технико-экономическое обоснование». Журнал медицинской химии. 52 (18): 5712–20. Дои:10.1021 / jm9006966. ЧВК 2745826. PMID 19719084.
- ^ Ли Х, Люнг К.С., Баллестер П.Дж., Вонг М.Х. (24 января 2014 г.). "istar: веб-платформа для крупномасштабной стыковки белок-лиганд". PLOS ONE. 9 (1): e85678. Bibcode:2014PLoSO ... 985678L. Дои:10.1371 / journal.pone.0085678. ЧВК 3901662. PMID 24475049.
- ^ а б Чжоу Х., Сколник Дж. (Январь 2013 г.). «FINDSITE (гребешок): подход к скринингу виртуальных лигандов в протеомном масштабе на основе потоков / структуры». Журнал химической информации и моделирования. 53 (1): 230–40. Дои:10.1021 / ci300510n. ЧВК 3557555. PMID 23240691.
- ^ Рой А., Сколник Дж. (Февраль 2015 г.). «LIGSIFT: инструмент с открытым исходным кодом для структурного выравнивания лигандов и виртуального скрининга». Биоинформатика. 31 (4): 539–44. Дои:10.1093 / биоинформатика / btu692. ЧВК 4325547. PMID 25336501.
- ^ Голтон А., Беллис Л.Дж., Бенто А.П., Чемберс Дж., Дэвис М., Херси А., Лайт И., МакГлинчи С., Михалович Д., Аль-Лазикани Б., Оверингтон Дж. П. (январь 2012 г.). «ChEMBL: крупномасштабная база данных по биоактивности для открытия лекарств». Исследования нуклеиновых кислот. 40 (Выпуск базы данных): D1100–7. Дои:10.1093 / нар / gkr777. ЧВК 3245175. PMID 21948594.
- ^ Вишарт Д.С., Нокс С., Гуо А.С., Шривастава С., Хассанали М., Стотхард П., Чанг З., Вулси Дж. (Январь 2006 г.). «DrugBank: всеобъемлющий ресурс для поиска и исследования лекарств in silico». Исследования нуклеиновых кислот. 34 (Выпуск базы данных): D668–72. Дои:10.1093 / nar / gkj067. ЧВК 1347430. PMID 16381955.
- ^ Валлах I, Хейфец А (2018). «Большинство критериев классификации на основе лигандов поощряют запоминание, а не обобщение». Журнал химической информации и моделирования. 58 (5): 916–932. arXiv:1706.06619. Дои:10.1021 / acs.jcim.7b00403. PMID 29698607.
- ^ Ирвин JJ (2008). «Тесты сообщества для виртуального просмотра». Журнал компьютерного молекулярного дизайна. 22 (3–4): 193–9. Bibcode:2008JCAMD..22..193I. Дои:10.1007 / s10822-008-9189-4. PMID 18273555. S2CID 26260725.
- ^ Хороший кондиционер, Oprea TI (2008). «Оптимизация методов CAMD. 3. Виртуальные исследования с обогащением: помощь или помеха в выборе инструмента?». Журнал компьютерного молекулярного дизайна. 22 (3–4): 169–78. Bibcode:2008JCAMD..22..169G. Дои:10.1007 / s10822-007-9167-2. PMID 18188508. S2CID 7738182.
- ^ Шнайдер Г (апрель 2010 г.). «Виртуальный просмотр: бесконечная лестница?». Обзоры природы. Открытие наркотиков. 9 (4): 273–6. Дои:10.1038 / nrd3139. PMID 20357802. S2CID 205477076.
- ^ Ballester PJ (январь 2011 г.). «Сверхбыстрое распознавание формы: метод и применение». Медицинская химия будущего. 3 (1): 65–78. Дои:10.4155 / fmc.10.280. PMID 21428826.
- ^ Лавеккья А., Ди Джованни С. (2013). «Стратегии виртуального скрининга в открытии лекарств: критический обзор». Современная лекарственная химия. 20 (23): 2839–60. Дои:10.2174/09298673113209990001. PMID 23651302.
- ^ Гуруджи К.С., Дешпанде В.Л. (февраль 1978 г.). «Улучшенный метод анализа каркаса». Компьютеры и конструкции. 8 (1): 147–152. Дои:10.1016/0045-7949(78)90171-2.
- ^ Прадипкиран, Джангампалли Ади; Редди, П. Хемачандра (март 2019 г.). «Структурно-ориентированный дизайн и исследования молекулярного докинга для фосфорилированных ингибиторов тау-белка при болезни Альцгеймера». Клетки. 8 (3): 260. Дои:10.3390 / ячеек8030260. ЧВК 6468864. PMID 30893872.
дальнейшее чтение
- Melagraki G, Afantitis A, Sarimveis H, Koutentis PA, Markopoulos J, Igglessi-Markopoulou O (май 2007 г.). «Оптимизация антагонистов рецептора MCH1 биарилпиперидина и 4-амино-2-биарилмочевины с использованием моделирования QSAR, методов классификации и виртуального скрининга». Журнал компьютерного молекулярного дизайна. 21 (5): 251–67. Bibcode:2007JCAMD..21..251M. Дои:10.1007 / s10822-007-9112-4. PMID 17377847. S2CID 19563229.
- Афантитис А, Мелаграки Дж., Саримвейс Х., Котентис П.А., Маркопулос Дж., Игглесси-Маркопулу О. (февраль 2006 г.). «Исследование влияния заместителя 1- (3,3-дифенилпропил) -пиперидинилфенилацетамидов на аффинность связывания CCR5 с использованием QSAR и методов виртуального скрининга». Журнал компьютерного молекулярного дизайна. 20 (2): 83–95. Bibcode:2006JCAMD..20 ... 83A. CiteSeerX 10.1.1.716.8148. Дои:10.1007 / s10822-006-9038-2. PMID 16783600. S2CID 21523436.
- Eckert H, Bajorath J (март 2007 г.). «Анализ молекулярного сходства в виртуальном скрининге: основы, ограничения и новые подходы». Открытие наркотиков сегодня. 12 (5–6): 225–33. Дои:10.1016 / j.drudis.2007.01.011. PMID 17331887.
- Уиллетт П. (декабрь 2006 г.). «Виртуальный скрининг на основе сходства с использованием 2D-отпечатков пальцев» (PDF). Открытие наркотиков сегодня (Представлена рукопись). 11 (23–24): 1046–53. Дои:10.1016 / j.drudis.2006.10.005. PMID 17129822.
- Фара, округ Колумбия, Опреа Т.И., Просниц Э.Р., Болога К.Г., Эдвардс Б.С., Скляр Л.А. (2006). «Интеграция виртуального и физического просмотра». Открытие лекарств сегодня: технологии. 3 (4): 377–385. Дои:10.1016 / j.ddtec.2006.11.003. ЧВК 7105924.
- Muegge I, Oloffa S (2006). «Успехи в виртуальном просмотре». Открытие лекарств сегодня: технологии. 3 (4): 405–411. Дои:10.1016 / j.ddtec.2006.12.002. ЧВК 7105922.
- Шнайдер Г (апрель 2010 г.). «Виртуальный просмотр: бесконечная лестница?». Обзоры природы. Открытие наркотиков. 9 (4): 273–6. Дои:10.1038 / nrd3139. PMID 20357802. S2CID 205477076.
внешняя ссылка
- VLS3D - список из более чем 2000 баз данных, онлайн и автономных in silico инструменты