Редукция Вольфа – Кишнера - Wolff–Kishner reduction
| Редукция Вольфа-Кишнера | |
|---|---|
| Названный в честь | Людвиг Вольф Николай Кишнер |
| Тип реакции | Органическая окислительно-восстановительная реакция |
| Идентификаторы | |
| Портал органической химии | Вольф-Кишнер-редукция |
| RSC ID онтологии | RXNO: 0000226 |
В Редукция Вольфа – Кишнера это реакция, используемая в органическая химия преобразовать карбонил функции в метиленовые группы. В контексте синтеза сложных молекул его наиболее часто используют для удаления карбонильной группы после того, как она выполнила свою синтетическую цель активации промежуточного соединения на предыдущей стадии. Таким образом, для этой реакции нет очевидного ретрона. Первоначально сообщил Николай Кишнер в 1911 г.[1] и Людвиг Вольф в 1912 г.,[2] он был применен для полного синтеза скопадульциновой кислоты B,[3] аспидоспермидин[4][5] и дисидиолид.[6]

В общем, механизм реакции в первую очередь включает на месте поколение гидразон путем конденсации гидразин с кетоновым или альдегидным субстратом. Однако иногда бывает выгодно использовать предварительно сформированный гидразон в качестве субстрата (см. модификации ). Стадия, определяющая скорость реакции, представляет собой депротонирование гидразона алкоксидным основанием с образованием диимид-аниона посредством согласованной стадии протонирования / депротонирования, опосредованной растворителем. Распад этого алкилдиимида с потерей N2 приводит к образованию алкиланиона, который может быть протонирован растворителем с получением желаемого продукта.

Поскольку восстановление Вольфа – Кишнера требует очень основных условий, оно не подходит для оснований, чувствительных к основанию. В некоторых случаях образование необходимого гидразона не будет происходить в стерически затрудненных карбонильных группах, что препятствует реакции. Однако этот метод может быть лучше, чем связанный с ним. Редукция Клемменсена для соединений, содержащих чувствительные к кислоте функциональные группы, такие как пирролы, и для высокомолекулярных соединений.
История
Редукция Вольфа – Кишнера была открыта независимо Н. Кишнером.[1] в 1911 г. и Людвиг Вольф в 1912 г.[2] Кишнер обнаружил, что добавление предварительно сформированных гидразон до горячего гидроксида калия, содержащего раздробленную платинированную пористую пластину, приводило к образованию соответствующего углеводорода. В 2013 году был опубликован обзор «Инвалидность, деспотизм, дезоксигенация - от ссылки до академика: Николай Матвеевич Кижнер», описывающий жизнь и деятельность Кишнера.[7]

Позже Вольф достиг того же результата, нагревая этанольный раствор семикарбазоны или же гидразоны в запаянной пробирке до 180 ° С в присутствии этилата натрия.
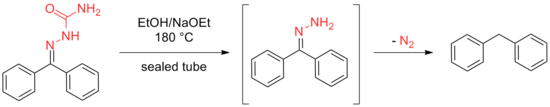
Преимущество метода, разработанного Кишнером, состоит в том, что он не требует наличия герметичной трубки, но обе методологии страдают ненадежностью при применении ко многим затрудненным субстратам. Эти недостатки способствовали развитию процедуры Вольфа, в которой использование высококипящих растворителей, таких как этиленгликоль и триэтиленгликоль были реализованы, чтобы обеспечить высокие температуры, необходимые для реакции, избегая при этом необходимости в герметичной пробирке.[8][9] За этими первоначальными модификациями последовало множество других улучшений, как описано ниже.
Механизм
Механизм редукции Вольфа – Кишнера был изучен Шмантом с соавторами.[10][11][12][13] Согласно исследованиям Шманта, первым шагом в этой реакции является образование аниона гидразона. 1 депротонированием концевого азота MOH. Если семикарбазоны используются в качестве субстратов, после первоначального превращения в соответствующий гидразон следует депротонирование.[2] Ряд механистических данных предполагает, что этап определения ставки включает образование новой углерод-водородной связи на углеродном конце в делокализованном гидразоновом анионе. Этот захват протона происходит согласованным образом с отводом второго протона на азотном конце, вызванного растворителем. Вывод Шманта о том, что эта реакция протекает первого порядка как по гидроксид-иону, так и по кетон-гидразону, поддерживает это механистическое предположение.[14] В этом процессе должны быть задействованы несколько молекул растворителя, чтобы обеспечить согласованный процесс. Подробный Анализ Хэммета[10] арилальдегидов, метиларилкетонов и диарилкетонов показали нелинейную зависимость, которую авторы приписывают сложности стадии определения скорости. Слегка электроноакцепторные заместители способствуют образованию углеродно-водородной связи, но сильно электроноакцепторные заместители уменьшают отрицательный заряд на концевом азоте и, в свою очередь, способствуют более крупной и твердой сольватной оболочке, которая затрудняет разрыв связи N-H. Наблюдаемая исключительно высокая отрицательная энтропия значений активации может быть объяснена высокой степенью организации в предлагаемом переходном состоянии.
Кроме того, было обнаружено, что скорость реакции зависит от концентрации гидроксильного растворителя и от катиона в алкоксидном катализаторе. Наличие краун-эфир в реакционной среде может увеличивать реакционную способность гидразон-аниона 1 за счет диссоциации ионной пары и, следовательно, увеличения скорости реакции.[13] Заключительным этапом восстановления Вольфа – Кишнера является коллапс диимид-аниона. 2 в присутствии источника протонов, чтобы получить углеводород за счет потери диазота, чтобы получить алкил-анион 3, который претерпевает быструю и необратимую кислотно-щелочную реакцию с растворителем с образованием алкана. Доказательства этого высокоэнергетического промежуточного продукта были получены Табером с помощью внутримолекулярного захвата. Стереохимический результат этого эксперимента больше соответствовал промежуточному алкил-аниону, чем альтернативной возможности алкильного радикала.[15] Общая движущая сила реакции - выделение газообразного азота из реакционной смеси.

Модификации
Многие усилия, направленные на улучшение восстановления Вольфа-Кишнера, были сосредоточены на более эффективном образовании промежуточного гидразона путем удаления воды и более высокой скорости разложения гидразона за счет увеличения температуры реакции.[8][9] Некоторые из новых модификаций обеспечивают более значительные улучшения и позволяют проводить реакции в значительно более мягких условиях. В таблице приведены краткие сведения о некоторых модификациях, которые были разработаны с момента первоначального открытия.
| Оригинальная процедура[1][2] | Хуанг Минлон[16] | Бартон[17] | Втиснуть[18] | Хенбест[19] | Калиоти[20] | Майерс[21] | |
|---|---|---|---|---|---|---|---|
| Реагенты | карбонильное соединение, 100% H2NNH2, Na или NaOEt | карбонильное соединение, 85% H2NNH2, КОН | карбонильное соединение, безводное H2NNH2, Na | предварительно сформированный гидразон, КОтБу | предварительно сформированный гидразон, КОтБу | тозилгидразон, донор гидрида | карбонильное соединение, 1,2-бис (терт-бутилдиметилсилил) - гидразин, Sc (OTf)3, КОтБу |
| Растворитель | высококипящий растворитель, например этиленгликоль | высококипящий растворитель, например этиленгликоль | высококипящий растворитель, например диэтиленгликоль гликоль | анх. ДМСО | толуол | THF | ДМСО |
| Температура | 200 ° С | 180–200 ° C (после удаления воды и избытка гидразина) | 210 ° С | 25 ° C | 111 ° С | 66 ° С | 25 ° С |
| Преимущества | одноэтапная процедура | сокращение времени реакции, возможность достижения более высоких температур, нет необходимости использовать anh. гидразин | позволяет декарбонилировать стерически затрудненные субстраты | протекает при комнатной температуре | не требуется медленное добавление гидразона | мягкие условия реакции, возможны с различными восстановителями | очень мягкие условия реакции |
| Недостатки | длительное время реакции (50–100 ч) | необходима перегонка | суровые условия реакции | необходимо выделение гидразона и медленное добавление | изоляция гидразона необходима | необходимо выделение тозилгидразона. донор гидрида может действовать как основание | синтез 1,2-бис (терт-бутилдиметилсилил) - гидразин необходим |
| Функциональная групповая толерантность | не переносит сложные эфиры, амиды, галогены, циано- и нитрогруппы | аналогично оригинальной процедуре | аналогично оригинальной процедуре | переносит амиды | более высокая переносимость α-заместители, которые подлежат устранению и α, β-ненасыщенные еноны, которые претерпят миграцию в исходных условиях | переносит сложные эфиры, амиды, циано-, нитро- и хлор-заместители с NaBH3CN как источник гидрида, не допускает первичных бром- и йодзаместителей | не сообщили |
Модификация Хуан Минлон
В 1946 г. Хуанг Минлон сообщили о модифицированной процедуре восстановления кетонов по Вольфу-Кишнеру, в которой избыток гидразина и воды удаляли перегонкой после образования гидразона.[16][22] Эффект понижения температуры воды, который был получен при образовании гидразона, обычно приводил к длительному времени реакции и суровым условиям реакции, даже если при образовании гидразона использовался безводный гидразин. Модифицированная процедура состоит из кипячения карбонильного соединения в 85% гидразингидрате с тремя эквивалентами гидроксида натрия с последующей отгонкой воды и избытка гидразина и повышением температуры до 200 ° C. Используя эту модификацию, можно получить значительно сокращенное время реакции и улучшенный выход. В первоначальном отчете Хуан Минлона описывается сокращение β-(п-феноксибензоил) пропионовой кислоты до γ-(п-феноксифенил) масляной кислоты с выходом 95% по сравнению с выходом 48%, полученным по традиционной методике.

Модификация Бартона
Через девять лет после первой модификации Хуанг Минлона Бартон разработал метод восстановления стерически затрудненных карбонильных групп.[17] Этот метод отличается строгим исключением воды, более высоких температур и более длительным временем реакции, а также натрия в диэтиленгликоль гликоль вместо алкоксидного основания. В этих условиях могут быть устранены некоторые проблемы, которые обычно возникают с затрудненными кетонами, например, C11-карбонильная группа в стероидном соединении, показанном ниже, успешно восстанавливалась в условиях Бартона, в то время как условия Хуанга-Минлона не влияли на это преобразование.

Заполнить модификацию
Медленное добавление предварительно образованных гидразонов к калию терт-бутоксид в ДМСО в качестве реакционной среды вместо гликолей позволяет успешно проводить образование углеводородов при температурах до 23 ° C.[18] Крам объяснил более высокую реакционную способность ДМСО как растворителя более высокой щелочной силой калия. терт-бутоксид в этой среде.

Эта модификация не использовалась в значительной степени в органическом синтезе из-за необходимости изолировать предварительно сформированные гидразоновые субстраты и добавлять гидразон в реакционную смесь в течение нескольких часов.
Модификация Хенбеста
Хенбест расширил процедуру Крама за счет кипячения карбонилгидразонов и калия. терт-бутоксид в сухом толуоле.[19] Медленное добавление гидразона не является необходимым, и было обнаружено, что эта процедура лучше подходит для карбонильных соединений, склонных к побочным реакциям, вызываемым основанием, чем модификация Крама. Например, было обнаружено, что миграция двойной связи в α, β-ненасыщенные еноны и устранение функциональных групп некоторых α-замещенные кетоны с меньшей вероятностью встречаются в условиях Хенбеста.[23]
Калиоти реакция
Лечение тозилгидразоны с гидрид-донорными реагентами для получения соответствующих алканов, известная как реакция Калиоти.[20][24] Изначально заявленные условия реакции были изменены, и доноры гидридов, такие как цианоборгидрид натрия, триацетоксиборгидрид натрия, или же катехолборан может восстанавливать тозилгидразоны до углеводородов.[25] Реакция протекает в относительно мягких условиях и поэтому может выдерживать более широкий набор функциональных групп, чем исходная процедура. Восстановление с цианоборгидридом натрия в качестве восстановителя можно проводить в присутствии сложных эфиров, амидов, циано-, нитро- и хлорзаместителей. Первичные бром- и йодзаместители замещаются нуклеофильным гидридом в этих условиях.

В нескольких статьях изучается механизм этого восстановления, и возможны множественные пути реакции, в зависимости от pH реакции, используемого восстанавливающего агента и электронных свойств субстрата. [26][27] Одна возможность, возникающая в кислых условиях, включает прямую гидридную атаку иминий ион 1 после предварительного протонирования тозилгидразона. Полученное производное тозилгидразина 2 впоследствии подвергается устранению п-толуолсульфиновая кислота и разлагается через диимин средний 3 к соответствующему углеводороду.

Небольшое изменение этого механизма происходит, когда таутомеризация к азогидразону способствует индуктивные эффекты. Переходный азогидразин 4 затем может быть восстановлен до производного тозилгидразина 2 и получают декарбонилированный продукт аналогично первому варианту. Этот механизм работает, когда используются относительно слабые доноры гидридов, такие как цианоборгидрид натрия. Известно, что цианоборгидрид натрия недостаточно силен для снижения имины, но может уменьшить иминий ионы.
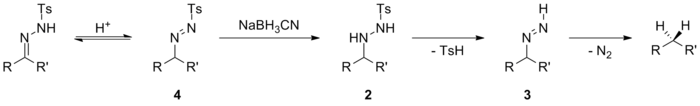
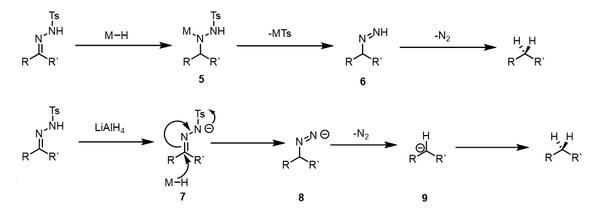
Когда используются более сильные доноры гидридов, действует другой механизм, который позволяет избежать использования кислых условий. Доставка гидрида происходит с образованием промежуточного 5, с последующим удалением металла сульфинат дать промежуточное азо 6. Этот промежуточный продукт затем разлагается с потерей газообразный азот, чтобы получить восстановленное соединение. Когда используются сильноосновные доноры гидридов, такие как литийалюминийгидрид, то депротонирование тозилгидразона может происходить до доставки гидрида. Промежуточный анион 7 может подвергаться гидридной атаке, удаляя сульфинат металла с образованием азоаниона 8. Это легко разлагается на карбанион 9, который протонируется с образованием восстановленного продукта.
Как и в случае исходного восстановления Вольфа-Кишнера, реакция декарбонилирования часто может не сработать из-за неудачного образования соответствующего тозилгидразона. Это типично для стерически затрудненных кетонов, как и в случае циклического аминокетона, показанного ниже.[28]

Если образование гидразона не дает образования, можно использовать альтернативные методы восстановления, в том числе тиокетал сокращение с Никель Ренея или реакция с триэтилборгидрид натрия.
Деоксигенация α, β-ненасыщенные карбонильные соединения
α, β-Ненасыщенные карбонилтозилгидразоны могут быть превращены в соответствующие алкены с миграцией двойной связи. Восстановление происходит стереоселективно, чтобы получить E геометрический изомер.[29]

Кабалка разработала очень мягкий метод. и другие. кто использовал один эквивалент катехолборан уменьшить α, β-ненасыщенные тозилгидразоны.[30]

Джерасси и другие. изучили механизм NaBH3Сокращение CN α, β-ненасыщенные тозилгидразоны. Основываясь на экспериментах по мечению дейтерием, они пришли к выводу, что образование алкена инициируется восстановлением гидрида иона иминия с последующей миграцией двойной связи и экструзией азота, которые происходят согласованным образом.[31]Аллильная диазеновая перегруппировка как заключительный этап восстановительной 1,3-транспозиции α, β-ненасыщенные тозилгидразоны до восстановленных алкенов также могут быть использованы для установления зр3-стереоцентры из аллильных диазенов, содержащие прохиральные стереоцентры. Влияние алкоксистереоцентра приводит к диастереоселективному восстановлению α, β-ненасыщенный тозилгидразон.[32] Авторы предсказали, что диастереоселективный перенос диазенового водорода на одну сторону прохирального алкена может быть усилен во время супрафациальной перегруппировки.

Модификация Майерса
В 2004 году Майерс и его коллеги разработали метод приготовления N-трет-бутилдиметилсилилгидразоны из карбонилсодержащих соединений.[21] Эти продукты могут быть использованы как превосходная альтернатива гидразонам при превращении кетонов в алканы. Преимуществами этой процедуры являются значительно более мягкие условия реакции и более высокая эффективность, а также удобство эксплуатации. Конденсация 1,2-бис (терт-бутилдиметилсилил) -гидразин с альдегидами и кетонами с Sc (OTf)3 как катализатор является быстрым и эффективным при температуре окружающей среды. Формирование и сокращение N-трет-бутилдиметилсилилгидразоны могут быть получены в однореакторном режиме с высоким выходом.

Недавно разработанный метод сравнивали непосредственно со стандартными условиями восстановления Хуанга – Минлона Вольфа – Кишнера (гидразингидрат, гидроксид калия, диэтиленгликоль, 195 ° C) для стероидного кетона, показанного выше. Продукт был получен с выходом 79% по сравнению с 91%, полученным при восстановлении через промежуточное соединение. N-трет-бутилдиметилсилилгидразон.
Побочные реакции
Восстановление Вольфа – Кишнера не подходит для чувствительных к основанию подложек и при определенных условиях может быть затруднено из-за стерическое препятствие окружающие карбонильную группу. Некоторые из наиболее распространенных побочных реакций перечислены ниже.
Формирование азина
Обычно встречающаяся побочная реакция при восстановлении Вольфа-Кишнера включает образование азина в результате реакции гидразона с карбонильным соединением. Образование кетона можно подавить, энергично исключив воду во время реакции. Некоторые из представленных процедур требуют выделения соединения гидразона перед восстановлением. Это может быть осложнено дальнейшим превращением гидразона продукта в соответствующий гидразин во время очистки продукта. Крам обнаружил, что образованию азина способствует быстрое добавление предварительно образованных гидразонов к калию. терт-бутоксид в безводном диметилсульфоксиде.[18]

Восстановление кетонов до спиртов этоксидом натрия
Вторая основная побочная реакция - это восстановление кетона или альдегида до соответствующего спирта. После начального гидролиза гидразона свободное карбонильное производное восстанавливается алкоксидом до карбинола. В 1924 году Эйзенлор сообщил, что значительные количества гидроксидекалина наблюдались во время попытки восстановления Вольфа-Кишнера транс-β-декалон.[33] Как правило, образование спирта можно подавить путем исключения воды или добавления избытка гидразина.
Устранение Кишнера – Леонарда
Кишнер отметил во время своих первоначальных расследований, что в некоторых случаях α-замещение карбонильной группы может привести к отщеплению с образованием ненасыщенных углеводородов в типичных условиях реакции. Позже Леонард развил эту реакцию и исследовал влияние различных α-заместители на исход реакции.[23][34] Он обнаружил, что степень выведения увеличивается с увеличением стерической массы уходящей группы. Более того, α-диалкиламинозамещенные кетоны обычно дают смесь продуктов восстановления и элиминирования, тогда как меньшее количество основных уходящих групп приводит к исключительному образованию алкенового продукта.

Фрагментация α, β-эпоксикетоны до аллиловых спиртов был расширен до синтетически полезного процесса и известен как Реакция Уортона.[35]
Расщепление или перегруппировка напряженных колец, прилегающих к карбонильной группе
Перегруппировка Гроба напряженных колец, прилегающих к карбонильной группе, наблюдали Эрман с соавторами.[36] Во время попытки Вольфа – Кишнера редукции транс-π-бромокамфора в условиях Крама, лимонен был выделен в качестве единственного продукта.
Точно так же может происходить разрыв напряженных колец, примыкающих к карбонильной группе. Когда 9β, 19-цикло-5α-прегнан-3,11,20-трион 3,20-диэтиленкеталь подвергали условиям Хуанга-Минлона, вместо образования 11-дезоксосоединения наблюдалось расширение кольца.[37]

Приложения
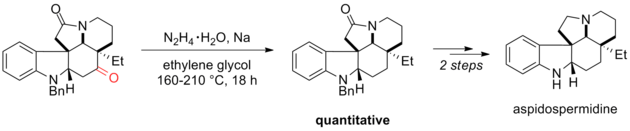
Восстановление Вольфа – Кишнера - эффективный инструмент в органическом синтезе. Например, Ishibashi и соавторы использовали модификацию Huang Minlon восстановления Вольфа – Кишнера в качестве одного из заключительных шагов в своем синтезе (±) -аспидоспермидина. Дистиллируемый материал удаляли после образования гидразона при 160 ° C, а затем нагревали до 210 ° C в течение ночи. Карбонильная группа, которая была восстановлена при восстановлении Вольфа-Кишнера, была важна для предыдущих стадий синтеза. Третичный амид был стабилен в условиях реакции и впоследствии был восстановлен алюмогидридом лития.[5]
Амиды обычно не являются подходящими субстратами для восстановления Вольфа-Кишнера, как показано в приведенном выше примере. Коу с соавторами, однако, обнаружили, что скрученный амид может быть эффективно восстановлен в условиях Вольфа-Кишнера.[38] Авторы объясняют это наблюдение стереоэлектронным смещением подложки, которое предотвращает «анти-Бредт ” иминий образование ионов и, следовательно, способствует выделению спирта и образованию гидразона. Функциональные группы амидов в этом напряженном субстрате можно рассматривать как изолированные функциональные группы аминов и кетонов, поскольку стабилизация резонанса предотвращается из-за торсионных ограничений. Продукт был получен с общим выходом 68% в двухстадийной методике.
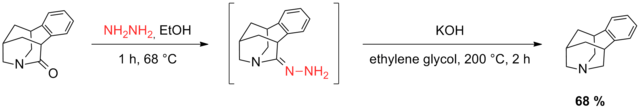
В 2011 году Петтус и Грин восстановили трициклическое карбонильное соединение, используя модификацию восстановления Вольфа – Кишнера Хуанг Минлон.[39] Несколько попыток декарбонилирования трициклического аллилацетата, содержащего кетон, потерпели неудачу, и пришлось удалить ацетатную функциональность, чтобы обеспечить успешное восстановление Вольфа-Кишнера. Наконец, аллиловый спирт был установлен с помощью кислородной канализации.

Восстановление Вольфа-Кишнера также использовалось в килограммах для синтеза функционализированного имидазольного субстрата. Было исследовано несколько альтернативных методов восстановления, но все испытанные условия остались безуспешными. Были учтены проблемы безопасности при крупномасштабном восстановлении по Вольфу-Кишнера, и высокооптимизированная процедура позволила получить продукт с хорошим выходом.[40]

Макинтош и другие. использовали перегруппировку аллильного диазена в своем синтезе C21–C34 фрагмент антаскомицина B.[41] Гидразон селективно восстанавливали катехолбораном, а избыток восстановителя разлагали тиосульфатом натрия. Затем неочищенный продукт реакции обрабатывали ацетатом натрия и нагревали с обратным холодильником в хлороформе с получением 1,4-син изомер.

Смотрите также
Рекомендации
- ^ а б c Кишнер, Н. (1911). «Редукция Вольфа – Кишнера; модификация Хуанга – Минлона». J. Russ. Phys. Chem. Soc. 43: 582–595.
- ^ а б c d Вольф, Л. (1912). "Chemischen Institut der Universität Jena: Methode zum Ersatz des Sauerstoffatoms der Ketone und Aldehyde durch Wasserstoff. [Erste Abhandlung.]". Annalen der Chemie Юстуса Либиха. 394: 86–108. Дои:10.1002 / jlac.19123940107.
- ^ Overman, L.E .; Ricca, D. J .; Тран, В. Д. (1993). «Первый полный синтез скопадульциновой кислоты B». Журнал Американского химического общества. 115 (5): 2042. Дои:10.1021 / ja00058a064.
- ^ Marino, J.P .; Rubio, M. B .; Cao, G .; Де Диос, А. (2002). «Полный синтез (+) - аспидоспермидина: новая стратегия энантиоспецифического синтеза алкалоидов аспидоспермы». Журнал Американского химического общества. 124 (45): 13398–13399. Дои:10.1021 / ja026357f. PMID 12418888.
- ^ а б Кавано, М .; Kiuchi, T .; Negishi, S .; Tanaka, H .; Хошикава, Т .; Matsuo, J. I .; Исибаши, Х. (2013). «Региоселективное меж- и внутримолекулярное формальное 4 + 2] циклоприсоединение циклобутанонов с индолами и полный синтез (±) -спидоспермидина». Angewandte Chemie International Edition. 52 (3): 906–10. Дои:10.1002 / anie.201206734. PMID 23184896.
- ^ Miyaoka, H .; Kajiwara, Y .; Hara, Y .; Ямада, Ю. (2001). «Полный синтез природного дисидиолида». Журнал органической химии. 66 (4): 1429–1435. Дои:10.1021 / jo0015772. PMID 11312976.
- ^ Льюис, Д. Э. (2013). «Инвалидность, деспотизм, дезоксигенация - от ссылки к академику: Кижнер Николай Матвеевич». Angewandte Chemie International Edition. 52 (45): 11704–11712. Дои:10.1002 / anie.201303165. PMID 24123691.
- ^ а б Herr, C.H .; Whitmore, F.C .; Шисслер, Р. В. (1945). «Реакция Вольфа-Кишнера при атмосферном давлении». Журнал Американского химического общества. 67 (12): 2061. Дои:10.1021 / ja01228a002.
- ^ а б Соффер, М. Д .; Соффер, М. Б .; Шерк, К. В. (1945). "Метод низкого давления для восстановления Вольфа-Кишнера". Журнал Американского химического общества. 67 (9): 1435. Дои:10.1021 / ja01225a004.
- ^ а б Szmant, H.H .; Хармут, К. М. (1964). «Реакция Вольфа-Кишнера гидразонов». Журнал Американского химического общества. 86 (14): 2909. Дои:10.1021 / ja01068a028.
- ^ Шмант, Х. Х. (1968). "Механизм реакций восстановления, отщепления и изомеризации Вольфа-Кишнера". Angewandte Chemie International Edition на английском языке. 7 (2): 120–128. Дои:10.1002 / anie.196801201.
- ^ Szmant, H.H .; Роман, М. Н. (1966). «Влияние диметилсульфоксида на скорость реакции Вольфа-Кишнера бензофенона гидразона1». Журнал Американского химического общества. 88 (17): 4034. Дои:10.1021 / ja00969a025.
- ^ а б Szmant, H.H .; Альчиатури, К. Э. (1977). «Механистические аспекты реакции Вольфа-Кишнера. 6. Сравнение гидразонов бензофенона, флуоренона, дибензотропона и дибензосуберона». Журнал органической химии. 42 (6): 1081. Дои:10.1021 / jo00426a034.
- ^ Szmant, H.H .; Harnsberger, H.F .; Батлер, Т. Дж .; Бари, У. П. (1952). "Кинетика реакции Вольфа-Кишнера диарилкетоновых гидразонов". Журнал Американского химического общества. 74 (11): 2724. Дои:10.1021 / ja01131a009.
- ^ Табер, Д. Ф .; Стачел, С. Дж. (1992). «О механизме редукции Вольфа-Кишнера». Буквы Тетраэдра. 33 (7): 903. Дои:10.1016 / S0040-4039 (00) 91571-5.
- ^ а б Хуанг-Минлон, [Н. А. (1946). «Простая модификация редукции Вольфа-Кишнера». Журнал Американского химического общества. 68 (12): 2487–2488. Дои:10.1021 / ja01216a013.
- ^ а б Osdene, T. S .; Тиммис, Г. М .; Maguire, M. H .; Shaw, G .; Goldwhite, H .; Saunders, B.C .; Clark, E. R .; Эпштейн, П. Ф .; Lamchen, M .; Стивен, А. М .; Tipper, C. F. H .; Eaborn, C .; Mukerjee, S.K .; Seshadri, T. R .; Willenz, J .; Робинсон, Р .; Thomas, A. F .; Hickman, J. R .; Kenyon, J .; Crocker, H.P .; Холл, Р. Х .; Burnell, R.H .; Taylor, W. I .; Watkins, W. M .; Barton, D.H.R .; Ives, D. A. J .; Томас, Б. Р. (1955). "Примечания". Журнал химического общества (возобновлено): 2038. Дои:10.1039 / JR9550002038.
- ^ а б c Cram, D. J .; Сахюн, М. Р. В. (1962). "Реакции восстановления Вольфа-Кишнера и элиминации при комнатной температуре". Журнал Американского химического общества. 84 (9): 1734. Дои:10.1021 / ja00868a048.
- ^ а б Grundon, M. F .; Henbest, H.B .; Скотт, М. Д. (1963). «344. Реакции гидразонов и родственных соединений с сильными основаниями. Часть I. Модифицированная процедура Вольфа-Кишнера». Журнал химического общества (возобновлено): 1855–1858. Дои:10.1039 / JR9630001855.
- ^ а б Caglioti, L .; Маги, М. (1963). «Реакция тозилгидразонов с алюмогидридом лития». Тетраэдр. 19 (7): 1127. Дои:10.1016 / S0040-4020 (01) 98571-0.
- ^ а б Furrow, M.E .; Майерс, А. Г. (2004). «Практические процедуры получения N-трет-бутилдиметилсилилгидразонов и их использование в модифицированных восстановлениях Вольфа-Кишнера и в синтезе галогенидов винила и гем-дигалогенидов». Журнал Американского химического общества. 126 (17): 5436–5445. Дои:10.1021 / ja049694s. PMID 15113215.
- ^ Хуанг-Минлон, [Н. А. (1949). «Восстановление стероидных кетонов и других карбонильных соединений по модифицированному методу Вольфа-Кишнера». Журнал Американского химического общества. 71 (10): 3301–3303. Дои:10.1021 / ja01178a008.
- ^ а б Леонард, Н. Дж .; Гельфанд, С. (1955). "Восстановление-устранение Кишнера. II. Α-Замещенные пинаколоны1,2". Журнал Американского химического общества. 77 (12): 3272. Дои:10.1021 / ja01617a036.
- ^ Калиоти, Л. (1966). «Восстановление тозилгидразонов и ацилтозилгидразидов». Тетраэдр. 22 (2): 487–493. Дои:10.1016/0040-4020(66)80015-7.
- ^ Hutchins, R.O .; Milewski, C.A .; Марьянов, Б. Э. (1973). «Селективная деоксигенация кетонов и альдегидов, включая затрудненные системы с цианоборгидридом натрия». Журнал Американского химического общества. 95 (11): 3662. Дои:10.1021 / ja00792a033.
- ^ Хатчинс, Р. О. (1991). Комп. Орг. Синтезатор. Пергамон. С. 327–362.
- ^ Миллер, В. П .; Yang, D. Y .; Weigel, T. M .; Han, O .; Лю, Х. В. (1989). «Исследования механистического разнообразия восстановления тозилгидразонов цианоборгидридом натрия». Журнал органической химии. 54 (17): 4175. Дои:10.1021 / jo00278a035.
- ^ Bosch, J .; Бонйох, Дж. (1981). «Синтетический путь к 6-функционализированным 2-азабицикло 3.3.1] нонанам». Журнал органической химии. 46 (8): 1538. Дои:10.1021 / jo00321a004.
- ^ Хатчинс, Р.O .; Качер, М .; Руа, Л. (1975). «Синтетическая полезность и механизм восстановительной деоксигенации & alpha;, & beta; -ненасыщенных п-тозилгидразонов с цианоборгидридом натрия». Журнал органической химии. 40 (7): 923. Дои:10.1021 / jo00895a024.
- ^ Кабалка, Г. З .; Ян, Д. Т. С .; Бейкер, Дж. Д. (1976). «Дезоксигенация альфа, бета-ненасыщенных альдегидов и кетонов посредством катехолборанового восстановления соответствующих тозилгидразонов». Журнал органической химии. 41 (3): 574. Дои:10.1021 / jo00865a043.
- ^ Тейлор, Э. Дж .; Джерасси, К. (1976). «Механизм восстановления цианоборгидридом натрия альфа, бета-ненасыщенных тозилгидразонов». Журнал Американского химического общества. 98 (8): 2275. Дои:10.1021 / ja00424a046.
- ^ Ци, Вт .; Макинтош, М.С. (2008). «Ациклический 1,4-стереоконтроль посредством восстановительных 1,3-транспозиций». Органические буквы. 10 (2): 357–359. Дои:10.1021 / ol702921x. ЧВК 2613761. PMID 18092798.
- ^ Eisenlohr, F .; Поленске, Р. (1924). "Über die raumisomeren Formen des Dekahydro-naphthalins (Dekalins)". Berichte der Deutschen Chemischen Gesellschaft (серии A и B). 57 (9): 1639. Дои:10.1002 / cber.19240570902.
- ^ Леонард, Н. Дж .; Гельфанд, С. (1955). "Восстановление-устранение Кишнера. I. Циклические и открытые цепи α-аминокетоны1,2". Журнал Американского химического общества. 77 (12): 3269. Дои:10.1021 / ja01617a035.
- ^ Wharton, P .; Болен, Д. (1961). «Связь - Восстановление гидразином α, β-эпоксидных кетонов до аллиловых спиртов». Журнал органической химии. 26 (9): 3615. Дои:10.1021 / jo01067a117.
- ^ Gustafson, D. H .; Эрман, У. Ф. (1965). «Новая фрагментация транс-π-бромкамфора». Журнал органической химии. 30 (5): 1665. Дои:10.1021 / jo01016a516.
- ^ Купчан, С. М .; Abushanab, E .; Шамасундар, К. Т .; Автор А. В. (1967). «Алкалоиды самшита. 13. Синтетический подход к системе 9 (10-19) абео-прегнан». Журнал Американского химического общества. 89 (24): 6327–6332. Дои:10.1021 / ja01000a060. PMID 6066048.
- ^ Bashore, C.G .; Самарджиев, И. Дж .; Bordner, J .; Коу, Дж. У. (2003). «Восстановление скрученного амида в условиях Вольфа-Кишнера: синтез производного бензо-1-аза-адамантана». Журнал Американского химического общества. 125 (11): 3268–3272. Дои:10.1021 / ja028152c. PMID 12630882.
- ^ Green, J.C .; Петтус, Т. Р. (2011). «Вызванный окислительной диароматизацией 5 + 2] каскад, обеспечивающий синтез α-цедрена, α-пипицола и сек-цедренола». Журнал Американского химического общества. 133 (5): 1603–1608. Дои:10.1021 / ja109925g. PMID 21194216.
- ^ Kuethe, J. T .; Чайлдерс, К. Г .; Peng, Z .; Журне, М .; Хамфри, Г. Р .; Викери, Т .; Bachert, D .; Лам, Т. Т. (2009). «Практическая реализация редукции Вольфа-Кишнера в масштабе килограмма». Исследования и разработки в области органических процессов. 13 (3): 576. Дои:10.1021 / op9000274.
- ^ Хатчисон, Джон М .; Гибсон, Эндрю С .; Уильямс, Дэвид Т .; Макинтош, Маттиас К. (2011). «Синтез C21 – C34 фрагмента антаскомицина B». Буквы Тетраэдра. 52 (48): 6349–6351. Дои:10.1016 / j.tetlet.2011.09.027. ISSN 0040-4039. ЧВК 3244276. PMID 22199407.
дальнейшее чтение
- Тодд, Д. Редукция Вольфа-Кишнера. В Орг. Реагировать. (ред. Адамс, Э.); John-Wiley & Sons, Inc .: Лондон, 1948 г., стр. 4, 378
- Хатчинс, Р.О. Восстановление C = X до CH2 Вольф-Кишнер и другие методы гидразона. В Комп. Орг. Synth. (ред. Трост, Б. М., Флеминг, И.); Пергамон: Оксфорд, 1991, стр.8, 327
- Льюис, Д. Э. Редукция Вольфа-Кишнера и связанные с ним реакции. Открытие и развитие; Эльзевир: Амстердам, 2019. ISBN-13 9780128157275].