ЧИП-экзо - ChIP-exo
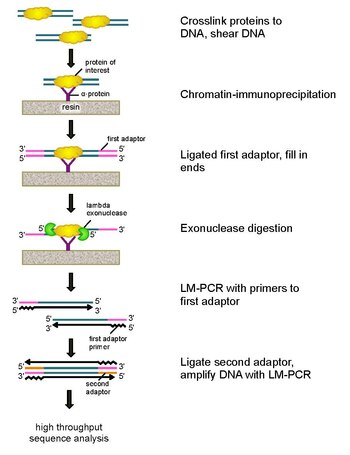
ЧИП-экзо это иммунопреципитация хроматина основанный на методе картирования местоположений интересующего белка (фактор транскрипции ) связывается с геномом. Это модификация ChIP-seq протокол, улучшающий разрешение сайтов привязки из сотен пар оснований почти до одной пары оснований. Он использует использование экзонуклеазы для разрушения цепей связанной с белком ДНК в направлении 5'-3 'с точностью до небольшого числа нуклеотидов сайта связывания белка. Нуклеотиды концов, обработанных экзонуклеазой, определяют с использованием некоторой комбинации Секвенирование ДНК, микрочипы, и ПЦР. Эти последовательности затем наносятся на карту генома, чтобы идентифицировать участки генома, в которых связывается белок.
Теория
Иммунопреципитация хроматина (ЧИП ) техники используются с 1984 г.[1] для обнаружения взаимодействий белок-ДНК. Для улучшения качества результатов на чипе было много вариантов. Одно такое улучшение, Чип-на-чипе (ChIP-chip), объединяет технологию ChIP с микрочипом. Этот метод имеет ограниченную чувствительность и специфичность, особенно in vivo где микроматрицы ограничены тысячами белков, присутствующих в ядерном компартменте, что приводит к высокому уровню ложноположительных результатов.[2] Далее пришел ChIP-секвенирование (ChIP-seq), который сочетает в себе ChIP с высокопроизводительным секвенированием.[3] Однако гетерогенная природа разрезанных фрагментов ДНК отображает сайты связывания с точностью до ± 300 пар оснований, что ограничивает специфичность. Во-вторых, загрязнение ДНК представляет собой серьезную проблему, поскольку так мало генетических локусов перекрестно связаны с интересующим белком, что делает любую неспецифическую геномную ДНК значительным источником фонового шума.[4]
Чтобы решить эти проблемы, Ри и Пью пересмотрели классическую анализ нуклеазной защиты разработать ChIP-exo.[5] Этот новый метод чипа основан на лямбда-выражении. экзонуклеаза который разрушает только и всю несвязанную двухцепочечную ДНК в 5'-3'-направлении. Вкратце, представляющий интерес белок (инженерный белок с эпитопной меткой может быть полезен для иммунопреципитации) сшивается in vivo с его естественными участками связывания в геноме с использованием формальдегида. Затем клетки собирают, разламывают, хроматин разрезают и растворяют с помощью обработка ультразвуком. Затем антитело используется для иммунопреципитации интересующего белка вместе со сшитой ДНК. Затем к концам лигируют адаптеры ДНК для ПЦР, которые служат начальной точкой для синтеза второй цепи ДНК после расщепления экзонуклеазой. Лямбда-экзонуклеаза затем переваривает двойные цепи ДНК с 5'-конца до тех пор, пока расщепление не блокируется на границе ковалентного взаимодействия белок-ДНК. Большая часть загрязняющих ДНК разрушается при добавлении второй однонитевой специфической экзонуклеазы. После сшивание в обратном порядке, праймеры к адаптерам ПЦР удлиняются с образованием двухцепочечной ДНК, а второй адаптер лигируется к 5'-концам, чтобы определить точное место прекращения расщепления экзонуклеазой. Затем библиотеку амплифицируют с помощью ПЦР, и продукты идентифицируют по высокопроизводительное секвенирование. Этот метод позволяет разрешить до одной пары оснований для любого сайта связывания белка в любом геноме, что является гораздо более высоким разрешением, чем ChIP-chip или ChIP-seq.
Преимущества
Было показано, что ChIP-exo уступает разрешению только одной пары оснований при идентификации мест связывания белка. Это контрастирует с ChIP-seq, который может определять место связывания белка только с ± 300 пар оснований.[4]
Загрязнение небелковых фрагментов ДНК может привести к высокому уровню ложноположительных и отрицательных результатов в экспериментах с ChIP. Добавление экзонуклеаз в процесс не только улучшает разрешение вызова сайта связывания, но и удаляет загрязняющую ДНК из раствора перед секвенированием.[4]
Белки, которые неэффективно связаны с нуклеотидным фрагментом, с большей вероятностью будут обнаружены ChIP-exo. Это позволило, например, узнать больше сайтов связывания факторов транскрипции CTCF, чем было обнаружено ранее.[5]
Из-за более высокого разрешения и меньшего фона при использовании ChIP-exo требуется меньшая глубина охвата секвенированием.[4]
Ограничения
Если комплекс белок-ДНК имеет несколько мест перекрестного сшивания в пределах одного события связывания, то может показаться, что существует несколько различных событий связывания. Это, вероятно, является результатом денатурирования этих белков и их перекрестного связывания в одном из доступных сайтов связывания в одном и том же событии. Затем экзонуклеаза остановится на одном из связанных сайтов, в зависимости от того, с каким сайтом сшивается белок.[5]
Как и в случае любого метода на основе ChIP, для использования этого метода необходимо наличие подходящего антитела к интересующему белку.
Приложения
Ри и Пью вводят ChIP-exo путем анализа небольшого набора факторов транскрипции: Reb1, Gal4, Phd1, Rap1 у дрожжей и CTCF у человека. Сайты Reb1 часто обнаруживались в кластерах, и эти кластеры имели в ~ 10 раз большую занятость, чем ожидалось. Вторичные сайты в кластерах были обнаружены на расстоянии ~ 40 п.н. от первичного сайта связывания. Связывающие мотивы Gal4 показали сильное предпочтение трем из четырех нуклеотидов, что свидетельствует об отрицательном взаимодействии между Gal4 и исключенным нуклеотидом. Phd1 распознает три разных мотива, что объясняет предыдущие сообщения о неоднозначности связывающего мотива Phd1. Было обнаружено, что Rap1 распознает четыре мотива. Гены рибосомных белков, связанные с этим белком, имели тенденцию использовать конкретный мотив с более сильной консенсусной последовательностью. Другие гены часто использовали кластеры более слабых консенсусных мотивов, возможно, для достижения аналогичной занятости. Связывающие мотивы CTCF задействовали четыре «модуля». Половина связанных сайтов CTCF использовала модули 1 и 2, а остальные использовали некоторую комбинацию из четырех. Считается, что CTCF использует свои цинковые пальцы для распознавания различных комбинаций этих модулей.[5]
Ри и Пью проанализировали структуру и организацию прединициативного комплекса (PIC) в Сахаромицеты геномы. Используя ChIP-exo, они смогли, среди других открытий, точно идентифицировать TATA-подобные функции в промоторах, которые, как сообщается, не имеют TATA.[6]
Смотрите также
Рекомендации
- ^ Гилмор, Д.С. Дж. Т. Лис (1983). "Обнаружение взаимодействий белок-ДНК in vivo: Распределение РНК-полимеразы по специфическим бактериальным генам ». Труды Национальной академии наук. 81 (14): 4275–4279. Дои:10.1073 / пнас.81.14.4275. ЧВК 345570. PMID 6379641.
- ^ Альберт, я; Т. Н. Маврич; LP Томшо; Дж Ци; SJ Zanton; СК Шустер; Б. Ф. Пью (2007). "Параметры трансляции и вращения нуклеосом H2A.Z пересекают Saccharomyces cerevisiae геном ". Природа. 446 (7135): 572–576. Bibcode:2007Натура.446..572А. Дои:10.1038 / природа05632. PMID 17392789. S2CID 4416890.
- ^ Рен, В; F Роберт; Джей Джей Уайрик; О Апарисио; Э. Дженнингс; Я Симон; J Zeitlinger; Дж. Шрайбер; Н. Ханнетт; E Kan; и другие. (2000). «Общегеномное расположение и функция ДНК-связывающих белков». Наука. 290 (5500): 2306–2309. Bibcode:2000Sci ... 290.2306R. CiteSeerX 10.1.1.123.6772. Дои:10.1126 / science.290.5500.2306. PMID 11125145.
- ^ а б c d Пью, Бенджамин. «Методы, системы и наборы для обнаружения взаимодействий белок-нуклеиновая кислота». Публикация заявки в США. Патенты США. Получено 17 февраля 2012.
- ^ а б c d Ри, Хо Сон; Би Джей Пью (2011). «Комплексные общегеномные взаимодействия белок-ДНК, обнаруженные при разрешении единичных нуклеотидов». Клетка. 147 (6): 1408–1419. Дои:10.1016 / j.cell.2011.11.013. ЧВК 3243364. PMID 22153082.
- ^ Ри, Хо Сон; Би Джей Пью (2012). «Полногеномная структура и организация прединициативных комплексов эукариот». Природа. 483 (7389): 295–301. Bibcode:2012Натура.483..295р. Дои:10.1038 / природа10799. ЧВК 3306527. PMID 22258509.
внешняя ссылка
- ДНК-белковые взаимодействия в высоком разрешении
- Разрешение связывания фактора транскрипции
- Иммунопреципитация хроматина высокого разрешения
- Важные белки регуляции генов, выявленные новым методом
- CexoR: пакет R / Bioconductor для выявления взаимодействий белок-ДНК с высоким разрешением в репликах ChIP-exo
- Peconic Genomics