Катализ водородной связью - Hydrogen-bond catalysis
Катализ водородной связью это тип органокатализ который полагается на использование водородная связь взаимодействия для ускорения и контроля органические реакции. В биологических системах водородные связи играют ключевую роль во многих ферментативных реакциях, как в ориентации молекул субстрата, так и в снижении барьеров для реакции.[1] Однако химики только недавно попытались использовать возможности использования водородных связей для проведения катализа, и эта область относительно неразвита по сравнению с исследованиями в Кислотный катализ Льюиса.[2]
Каталитические количества доноров водородных связей могут способствовать реакциям посредством множества различных механизмов. В ходе реакции водородная связь может использоваться для стабилизации анионных промежуточных продуктов и переходные состояния. В качестве альтернативы некоторые катализаторы могут связывать небольшие анионы, обеспечивая образование реактивных электрофильных катионов. Более кислые доноры могут действовать как общие или специфические кислоты, которые активируют электрофилы путем протонирования. Мощный подход - одновременная активация обоих партнеров в реакции, например нуклеофил и электрофил, называемые «бифункциональным катализом». Во всех случаях тесная ассоциация молекулы катализатора с субстратом также делает катализ водородными связями мощным методом индукции энантиоселективность.
Катализаторы водородного связывания часто просты в изготовлении, относительно надежны и могут быть синтезированы с высокой энантиомерной чистотой. Новые реакции, катализируемые донорами водородных связей, обнаруживаются все более быстрыми темпами, включая асимметричные варианты обычных органических реакций, полезных для синтеза, такие как альдол дополнения, Дильс-Альдер циклоприсоединения и Манних реакции.[3]
Однако есть несколько проблем, которые необходимо преодолеть, прежде чем катализ на водородных связях сможет полностью раскрыть свой потенциал с точки зрения синтетической полезности. Известные в настоящее время реакции очень специфичны для субстрата и обычно имеют низкую скорость ускорения и оборота, что требует высокой загрузки катализатора. Катализаторы часто обнаруживаются и оптимизируются методом проб и ошибок, а химики плохо понимают взаимосвязь между структурой катализатора и реакционной способностью. Кроме того, эта область страдает отсутствием общего механистического понимания, которое значительно опередило открытие новых реакций. С более детальным изучением структуры и механизма в будущем, катализ водородных связей имеет большой потенциал для создания новых, эффективных, селективных реакций и многообещающих применений в асимметричном синтезе.
Каталитические стратегии
Стабилизация тетраэдрических интермедиатов
Многие полезные органические реакции включают образование тетраэдрические промежуточные соединения посредством нуклеофильной атаки функциональных групп, таких как альдегиды, амиды или же имины. В этих случаях катализ с использованием доноров водородных связей является привлекательной стратегией, поскольку анионные тетраэдрические промежуточные соединения являются лучшими акцепторами водородных связей, чем исходное соединение. Это означает, что по сравнению с исходным комплексом катализатор-подложка стабилизируется переходное состояние, несущее более отрицательный заряд.

Например, в типичной реакции ацильного замещения исходное карбонильное соединение координируется с катализатором через одну, две или, возможно, несколько водородных связей. Во время атаки нуклеофила на кислороде накапливается отрицательный заряд, пока не будет достигнут тетраэдрический промежуточный продукт. Следовательно, формально отрицательный кислород участвует в гораздо более сильной водородной связи, чем исходный карбонильный кислород из-за его повышенного отрицательного заряда. С энергетической точки зрения это приводит к снижению промежуточного и переходное состояние, тем самым ускоряя реакцию.
Этот способ катализа находится в активные сайты из многих ферменты, такой как сериновые протеазы.[4] В этом примере карбонил амида координирован с двумя донорами N – H. Эти участки множественной координации, предназначенные для ускорения карбонильных реакций в биологии, называются "оксианионные дыры Доставка серинового нуклеофила образует тетраэдрический промежуточный продукт, который стабилизируется увеличением водородной связи с оксианионным отверстием.

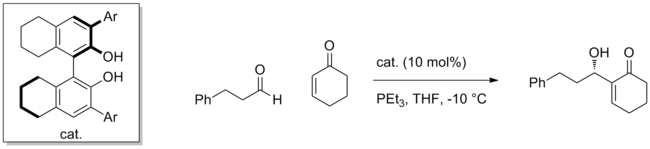
Многие синтетические катализаторы смогли успешно использовать эту стратегию для активации различных электрофилов. Использование хирального БИНОЛ катализатор, например, Реакция Морита-Бейлиса-Хиллмана добавление енонов к альдегидам может осуществляться с высокой энантиоселективностью.[5] Нуклеофил представляет собой разновидность енолятного типа, полученную в результате конъюгированного добавления PEt3 к енону и энантиоселективно добавляет к альдегиду, координированному с катализатором.
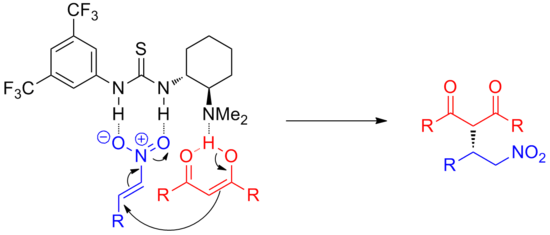
Помимо карбонилов, могут успешно использоваться другие электрофилы, такие как имины. Например, при использовании простого хирального тиомочевинного катализатора асимметричный Реакция Манниха ароматических иминов с силилкетенацеталями можно катализировать с высоким э.и. при почти количественном превращении.[6] Механизм этой реакции полностью не выяснен, и реакция очень специфична для субстрата, эффективна только для определенных ароматических электрофилов.
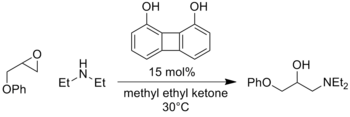
Возможности этого способа активации огромны, с постоянными новыми сообщениями о различных комбинациях электрофилов, нуклеофилов и структур катализатора. Кроме того, аналогичные реакции с участием оксианионных промежуточных продуктов, таких как присоединение енолята к нитрозо соединения[7] или открытие эпоксиды[8] также были успешно катализированы с помощью этой стратегии.
Однако, несмотря на количество известных различных реакций, общее понимание способа катализа ограничено, и почти все обнаруженные реакции чрезвычайно специфичны для субстрата.
Стабилизация анионных фрагментов
Другая стратегия, которая была исследована, - это стабилизация реакций, которые приводят к развитию частичных отрицательных зарядов в переходном состоянии. Примеры успешных применений - это, как правило, реакции, близкие к согласованным и перициклическим по природе. В ходе реакции один фрагмент приобретает частичный отрицательный характер, и переходное состояние можно стабилизировать, приняв водородную связь (и).
Наглядный пример - катализ Клейзеновские перестановки сложноэфирзамещенных аллилвиниловых эфиров, сообщенных исследовательской группой Якобсена.[9] Хиральный гуанидиний Было обнаружено, что катализатор успешно ускоряет реакцию при температуре около комнатной с высокой энантиоселективностью. Во время переходного состояния фрагмент, координированный с амидиниевым катализатором, приобретает частичный анионный характер из-за электроотрицательности кислорода и электроноакцепторной сложноэфирной группы. Это увеличивает прочность водородных связей и снижает энергию переходного состояния, тем самым ускоряя реакцию.

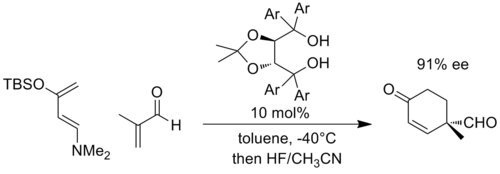
Точно так же отрицательный заряд может развиваться в реакциях циклоприсоединения, таких как Дильс-Альдер реакция, когда партнеры заменены соответствующим образом. В качестве типичного примера Равал и его коллеги разработали хиральный катализатор на основе α, α, α, α-тетраарил-1,3-диоксолан-4,5-диметанола (ТАДДОЛ ), которые могут катализировать реакции Дильса-Альдера. В следующем примере считается, что реакция с диеном с высоким содержанием электронов и диенофилом с низким содержанием электронов приводит к развитию значительного отрицательного заряда на енальном фрагменте, и если переходное состояние стабилизируется за счет увеличения водородной связи с TADDOL (Ar = 1 -нафтил).[10]
Связывание анионов
Катализаторы с водородной связью также могут ускорять реакции, способствуя образованию электрофильных частиц путем выделения и / или координации аниона, такого как галогенид. Катализаторы на основе мочевины и тиомочевины являются наиболее распространенными донорами в анион-связывающем катализе, и их способность связывать галогениды и другие анионы хорошо известна в литературе.[11] Использование хиральных анион-связывающих катализаторов может создать асимметричную ионную пару и вызвать замечательную стереоселективность.
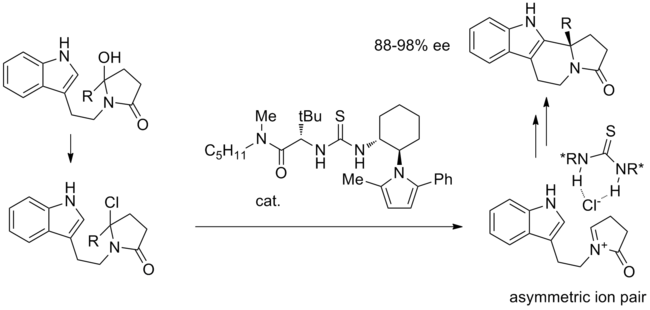
Одной из первых реакций, предложенных для проведения анион-связывающего катализа, является реакция Пикте-Шпенглер циклизация гидроксилактамов с TMSCl при тиомочевинном катализе.[12] В предлагаемом механизме после первоначального замещения гидроксильной группы на хлорид образуется пара ключевых ионов. Активированный иминиевый ион тесно связан с хиральным хлоридом, связанным с тиомочевиной, и внутримолекулярная циклизация протекает с высокой стереоселективностью.
Асимметричные ионные пары также могут быть атакованы в межмолекулярных реакциях. В интересном примере асимметричное присоединение енолсилановых нуклеофилов к ионам оксокарбения может быть осуществлено каталитическим образованием оксокарбения посредством связывания анионов.[13] Исходя из ацеталя, хлорэфир образуется с трихлорид бора и реагировал с енольным силаном и катализатором. Механизм образования комплекса оксокарбений-тиомочевина-хлорид полностью не выяснен. Считается, что в условиях реакции хлорэфир может эпимеризоваться, а тиомочевина может стереоселективно связывать хлорид с образованием тесно связанной ионной пары. Затем эта асимметричная ионная пара подвергается атаке силана с образованием алкилированного продукта.

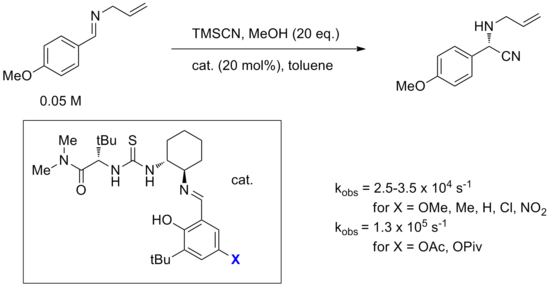
Ярким примером механизма связывания анионов является гидроцианирование иминов, катализируемое амидотиомочевинным катализатором Якобсена, изображенным на диаграмме ниже. Эта реакция также является одной из наиболее широко изученных с помощью вычислительных, спектроскопических, меченых и кинетических экспериментов.[14] Хотя рассматривалось прямое добавление цианида к имину, связанному с катализатором, альтернативный механизм, включающий образование пары имин-цианид, контролируемой катализатором, был рассчитан таким образом, чтобы барьер был ниже на 20 ккал / моль. Предлагаемый наиболее вероятный механизм начинается со связывания катализатора с HNC, который находится в равновесии с HCN. Этот комплекс затем протонирует молекулу имина, образуя ионно-цианидную ионную пару со связыванием катализатора и стабилизацией цианид-аниона. Считается, что иминиум также взаимодействует с амид-карбонилом на молекуле катализатора (см. Бифункциональный катализ ниже). Связанный цианид-анион затем вращается и атакует иминий через углерод. Исследователи пришли к выводу, что, хотя связывание имина и мочевины наблюдали с помощью спектроскопии и подтверждали ранние кинетические эксперименты, связывание имина происходит вне цикла, и все доказательства указывают на этот механизм с участием цианида, связанного с тиомочевиной.

Протонирование
Часто бывает трудно отличить катализ водородными связями от общий кислотный катализ.[3] Доноры водородной связи могут иметь различную кислотность, от слабой до по существу сильной кислоты Бренстеда, такой как фосфорная кислота. Глядя на степень переноса протона в ходе реакции, сложно, и в большинстве реакций она не была тщательно исследована. Тем не менее, сильнокислотные катализаторы часто группируются с катализаторами с водородными связями, поскольку они представляют крайность в этом континууме, и их каталитическое поведение имеет сходство. Механизм активации этих реакций включает начальное протонирование электрофильного партнера. Это приводит к тому, что подложка становится более электрофильной и создается ионная пара, через которую можно передавать стереохимическую информацию.

Асимметричный катализ, включающий почти полное протонирование субстрата, был эффективен в реакциях Манниха ароматических альдиминов с углеродными нуклеофилами.[15] Кроме того, аза-Реакции Фриделя-Крафтса из фураны, амидоалкилирование из диазокарбонил соединения, асимметричные гидрофосфонилирование из альдимины и перенос гидрирования также поступали сообщения.[3] Хиральные кислоты Бренстеда часто легко получить из хиральных спиртов, таких как BINOL, и многие из них уже присутствуют в литературе из-за их признанной полезности в исследованиях молекулярного распознавания.[16]
Многофункциональные стратегии
Одним из основных преимуществ катализа водородными связями является способность создавать катализаторы, которые участвуют во множественных нековалентных взаимодействиях для ускорения реакции. В дополнение к использованию доноров водородных связей для активации или стабилизации реакционного центра во время реакции, можно ввести другие функциональные группы, такие как Базы Льюиса, арены или участки присоединения водородных связей для придания дополнительной стабилизации или для влияния на другого реактивного партнера.
Например, природный фермент хоризмат мутаза, который катализирует перегруппировку Клайзена хоризмата, обладает множеством других взаимодействий в дополнение к водородным связям, участвующим в стабилизации енолатоподобного фрагмента, что является примером стратегии стабилизации анионного фрагмента, описанной выше.[17] Ключевым взаимодействием является стабилизация другого катионного аллил фрагмент через взаимодействие катион-пи в переходном состоянии. Использование многих дополнительных водородных связей преследует несколько предполагаемых целей. Стабилизация множественных водородных связей с ферментом помогает преодолеть энтропийную стоимость связывания. Кроме того, взаимодействия помогают удерживать субстрат в реактивной конформации, а реакция, катализируемая ферментами, имеет энтропию активации, близкую к нулю, в то время как типичные перегруппировки Клейзена в растворе имеют очень отрицательную энтропию активации.

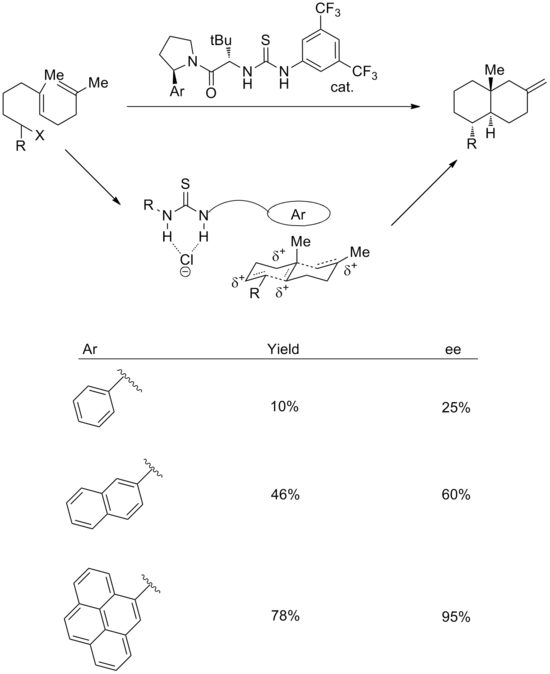
Использование катион-пи-взаимодействий также успешно реализовано в реакциях с синтетическими катализаторами. Комбинация стратегий связывания анионов и катион-pi может использоваться для осуществления энантиоселективной катионной полициклизации.[18] Предполагается, что в переходном состоянии тиомочевинная группа связывает хлорид, а ароматическая система стабилизирует связанный полиеновый катион. В подтверждение этого увеличение размера ароматического кольца приводит к улучшению как выхода, так и стереоселективности. Энантиоселективность хорошо коррелирует как с поляризуемостью, так и с квадрупольным моментом арильной группы.
Поскольку такое большое количество катализаторов и реакций включает связывание с электрофилами для стабилизации переходного состояния, многие бифункциональные катализаторы также содержат основную Льюиса, акцепторную водородную связь. В качестве типичного примера Дэн и его коллеги разработали тиомочевинно-аминный катализатор, способный стимулировать стереоселективные реакции Михаэля.[19] В предложенном переходном состоянии один из доноров тиомочевины N – H координирован с акцептором Михаэля и будет стабилизировать накопление отрицательного заряда. Основная неподеленная пара азота действует как акцептор водородной связи для координации нуклеофила, но в переходном состоянии действует как общее основание, способствующее присоединению нуклеофильного енолята.

Этот мотив вовлечения в реакцию как нуклеофильных, так и электрофильных партнеров и их стабилизации в переходном состоянии очень распространен в бифункциональном катализе, и многие другие примеры можно найти в статье о органокатализ тиомочевины.
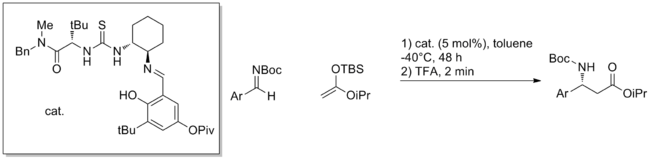
Относительно новая стратегия использования синтетических олигопептидов для проведения катализа дала множество успешных примеров каталитических методов.[20] Пептиды обладают множеством потенциальных сайтов для водородных связей, и обычно непонятно, как они взаимодействуют с субстратом или как они способствуют реакции. Преимущество пептидов состоит в том, что они чрезвычайно модульны, и часто эти катализаторы просеиваются большими массивами. Таким образом были обнаружены высокоэнантиоселективные реакции, такие как альдольная реакция, изображенная ниже.
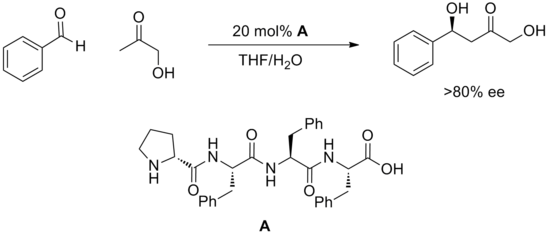
Другие превращения, успешно катализируемые синтетическими пептидами, включают гидроцианирование, ацилирование, присоединение конъюгата, альдегид-иминное связывание, альдольную реакцию и бромирование. Хотя природа переходных состояний неясна, во многих примерах небольшие изменения в структуре катализатора оказывают драматическое влияние на реактивность. Предполагается, что большое количество водородных связей как внутри пептида, так и между катализатором и субстратом должно взаимодействовать, чтобы соответствовать геометрическим требованиям для успешного катализа. Помимо этого, понимание конструкции и механизма катализатора еще не продвинулось дальше необходимости тестирования библиотек пептидов.
Дизайн катализатора
Привилегированные структуры
Типы доноров водородных связей, используемых в катализе, широко варьируются от реакции к реакции, даже среди аналогичных каталитических стратегий. Хотя конкретные системы часто тщательно изучаются и оптимизируются, общее понимание оптимального донора для реакции или взаимосвязи между структурой катализатора и реакционной способностью сильно отсутствует. Пока еще нецелесообразно рационально проектировать структуры, способствующие желаемой реакции с желаемой селективностью. Однако современный катализ водородных связей в первую очередь сосредоточен на нескольких типах систем, которые экспериментально кажутся эффективными в самых разных ситуациях.[21] Их называют «привилегированными структурами». Однако стоит отметить, что другие структурные каркасы и мотивы также показали многообещающие результаты, такие как координированные металлами доноры водородных связей.[22]
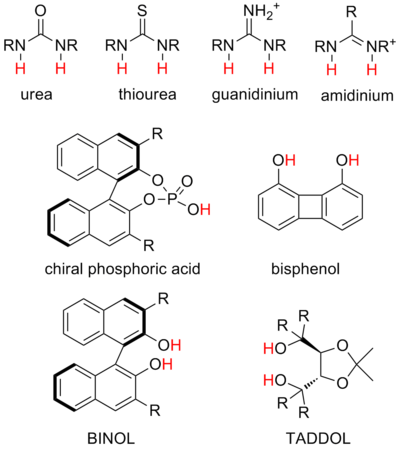
- Мочевина и тиомочевины являются наиболее распространенными структурами и могут стабилизировать различные отрицательно заряженные промежуточные соединения, а также участвовать в анион-связывающем катализе. Бифункциональный катализ мочевиной и тиомочевиной широко представлен в литературе.
- Гуанидиний и амидиний ионы являются структурными родственниками мочевины и тиомочевины и могут катализировать аналогичные реакции, но в силу своего положительного заряда являются более сильными донорами и намного более кислыми. Считается, что механизм гуанидиниевого и амидиниевого катализа часто включает частичное протонирование субстрата.
- Диол Считается, что катализаторы взаимодействуют с субстратом с помощью одинарной водородной связи, а другой гидроксил участвует во внутренней водородной связи. Это одни из первых исследованных катализаторов водородной связи. Они чаще всего используются для стабилизации частичного анионного заряда в переходных состояниях, например, для координации с диенофилами альдегидов в гетеро-реакциях Дильса-Альдера.
- Фосфорная кислота Катализаторы являются наиболее распространенными сильнокислотными катализаторами и работают путем образования хиральных ионных пар с основными субстратами, такими как имины.
Настройка катализатора
В целом кислотность донорских сайтов хорошо коррелирует с силой донора. Например, распространенной стратегией является добавление электроноакцепторных арильных заместителей к тиомочевинному катализатору, что может повысить его кислотность и, следовательно, силу его водородных связей. Однако до сих пор неясно, как сила донора коррелирует с желаемой реакционной способностью. Важно отметить, что более кислотные катализаторы не обязательно более эффективны. Например, мочевины менее кислые, чем тиомочевины, примерно на 6 единиц pKa, но в целом неверно, что мочевины значительно хуже катализируют реакции.[23]
Более того, влияние различных заместителей на катализатор редко бывает хорошо изучено. Небольшие замены заместителя могут полностью изменить реакционную способность или селективность. Примером этого были исследования оптимизации бифункционального катализатора реакции Штрекера, одного из первых хорошо изученных тиомочевинных катализаторов.[24] В частности, варьируя заместитель X на салицилальдиминовом заместителе, было обнаружено, что типичные электроноакцепторные или электронодонорные заместители мало влияют на скорость, но сложноэфирные заместители, такие как ацетат или пивалоат, по-видимому, вызывают заметное ускорение скорости. Это наблюдение трудно рационализировать, учитывая, что группа X находится далеко от реактивного центра в ходе реакции, и электроника не кажется причиной. В целом, несмотря на относительную простоту электронной настройки с помощью органических катализаторов, химики еще не достигли полезного понимания этих модификаций.
Синтетические приложения
Синтез натурального продукта
На сегодняшний день существует несколько примеров катализа водородными связями в синтезе природных продуктов, несмотря на большое количество обнаруженных реакций. Как правило, при высокой требуемой загрузке катализатора и часто экстремальной специфичности к субстрату катализ водородными связями еще не разработан в достаточной степени, чтобы обеспечить полезные общие реакции, которые представляют собой значительное улучшение по сравнению с традиционными методами. В опубликованных примерах катализ водородными связями в основном используется на начальных стадиях для быстрого доступа к ранним промежуточным продуктам с высоким энантиомерным обогащением.
В синтезе Якобсена (+) - йохимбина,[25] индоловый алкалоид, ранняя энантиоселективная реакция Пикте-Шпенглера с использованием пирролзамещенного тиомочевинного катализатора дала количество продукта в граммовых количествах с ее э.и. 94% и выходом 81%. Остаток синтеза был коротким с использованием восстановительного аминирования и внутримолекулярной реакции Дильса-Альдера.

В 2008 году Такемото раскрыл краткий синтез (-) - эпибатидина, основанный на каскаде Майкла, катализируемом бифункциональным катализатором.[26] После первоначального асимметричного добавления Майкла к β-нитростирол внутримолекулярное добавление Михаэля дает циклический кетоэфир с 75% ее. Стандартные манипуляции с функциональными группами и внутримолекулярная циклизация дают натуральный продукт.

Масштабируемый синтез строительных блоков
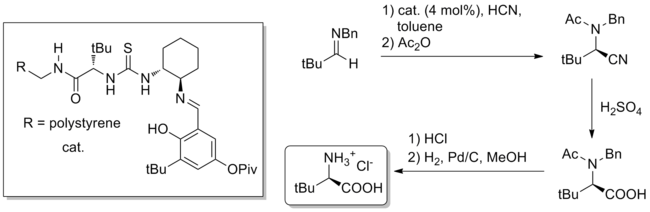
Помимо полного синтеза, потенциально полезным применением катализа водородных связей является массовый синтез труднодоступных хиральных небольших молекул. Ярким примером является граммовая шкала. Синтез Стрекера неестественного аминокислоты с использованием тиомочевинного катализа, сообщается в журнале Природа в 2009.[27] Катализатор, связанный с полимером или гомогенный, является производным природного трет-лейцин и может катализировать (загрузка катализатора 4 мол.%) образование продукта Штрекера из бензгидриламинов и водной HCN. Гидролиз нитрила и снятие защиты дает чистый неприродный трет-лейцин с общим выходом 84% и ее э.и. 99%.
Проблемы и цели на будущее
Несмотря на широко распространенный интерес к органокатализу и большое количество новых каталитических систем, которые постоянно открываются, прогресс в понимании механизма и конструкции катализатора в области катализа водородными связями крайне ограничен. По сравнению с более развитой областью, такой как реакции сочетания, катализируемые палладием Катализ на основе водородных связей представляет собой множество проблем, которые еще не решены.
- Оборот: Хотя реакции, катализируемые палладием, часто могут быть эффективными при загрузке катализатора менее 0,1 мол.%, Катализаторы с водородной связью часто добавляют в количестве более 10 мол.%. Плохое ускорение скорости - это общая тенденция, которую необходимо преодолеть, чтобы катализ на водородных связях стал практической синтетической стратегией.
- Механизм: В будущем потребуются дальнейшие исследования точных шагов, вовлеченных в механизм катализа водородных связей, что позволит химикам рационально разрабатывать каталитические стратегии для более сложных или более полезных превращений. Для сравнения: основные этапы кросс-сочетания, катализируемого палладием, систематически и тщательно изучались в течение последних нескольких десятилетий и привели к значительному прогрессу в области каталитического охвата, контроля и принципов построения реакций. Например, улучшенное понимание окислительная добавка привело к тому, что арилхлориды стали практическими партнерами по связыванию, в то же время улучшив понимание восстановительное устранение привело к развитию новых реакций с участием зр.3 центры. Зная эти фундаментальные каталитические стадии, способность рационально планировать новые реакции и каскады оказалась чрезвычайно полезной в области полного синтеза. Напротив, нам не хватает общего систематического механистического понимания стадий катализа водородных связей и того, как на них влиять. Подробные механистические исследования до сих пор ограничивались отдельными системами, и их результаты не имели очевидного прогностического использования.
- Катализатор: Связанная проблема - исследование того, как изменения в катализаторе, структурных, конформационных и электронных могут быть использованы для рационального влияния на реакцию. Цель состоит в том, чтобы полностью понять, как использовать несколько совместных взаимодействий, чтобы максимально ускорить реакцию и придать селективность. В идеале рациональная конструкция катализатора в конечном итоге заменит скрининг семейств катализаторов, и выбор строительных блоков станет более систематическим.
- Объем: В то время как новые реакции постоянно открываются, большинство реакций имеют чрезвычайно узкую область применения субстрата, и причина такой узкой области часто не понимается. В области палладиевого катализа после того, как были установлены основы механистического понимания, количество реакций стало быстро расти. Знание факторов, влияющих на каждую стадию катализа, позволило химикам предвидеть и проводить новые высокоэффективные синтетические реакции, такие как реакции активации связи C-H. В области катализа водородных связей химики еще не достигли стадии, на которой можно было бы легко и систематически определять новые типы реакционной способности. На этом этапе открытие реакции полезно, но требуется более детальное изучение механизмов, чтобы реализовать весь потенциал катализа водородных связей.
Смотрите также
дальнейшее чтение
- Катализ водородными связями. Презентация собрания Evans Group Питера Х. Фуллера. Связь
- Асимметричный катализ водородных связей. Презентация встречи MacMillan Group Энтони Мастраккио. Связь
- Водородная связь в асимметричном катализе. Презентация встречи Leighton Group Уттама Тамбара. Связь
- Асимметричный катализ хиральными донорами водородных связей. Презентация встречи Wipf Group от Чжэнлай Фана Связь
- Энантиоселективный органокатализ. Эд. Питер И. Далко, Wiley-VCH: Weinheim, 2007.
Рекомендации
- ^ Jacobsen, E. N .; Ноулз Р. Р. (сентябрь 2010 г.). «Привлекательные нековалентные взаимодействия в асимметричном катализе: связи между ферментами и низкомолекулярными катализаторами» (PDF). Proc. Natl. Акад. Наука. 107 (48): 20678–20685. Bibcode:2010PNAS..10720678K. Дои:10.1073 / pnas.1006402107. ЧВК 2996434. PMID 20956302.
- ^ Jacobsen, E. N .; Тейлор, М.С. (февраль 2006 г.). «Асимметричный катализ хиральными донорами водородной связи». Энгью. Chem. Int. Эд. 45 (10): 1521–1539. Дои:10.1002 / anie.200503132.
- ^ а б c Дойл, Эбигейл Дж .; Якобсен, Э. Н. (декабрь 2007 г.). «Низкомолекулярные доноры водородной связи в асимметричном катализе». Chem. Rev. 107 (12): 5713–5743. Дои:10.1021 / cr068373r. PMID 18072808.
- ^ Синнотт, М. (1998). Комплексный биологический катализ, Vol. 1. Лондон: Academic Press. С. 345–379.
- ^ McDougal, N.T .; Шаус, С. Э. (сентябрь 2003 г.). «Асимметричные реакции Морита-Бейлиса-Хиллмана, катализируемые хиральными кислотами Бренстеда». Варенье. Chem. Soc. 125 (40): 12094–12095. Дои:10.1021 / ja037705w. PMID 14518986.
- ^ Wenzel, A. G .; Якобсен, Э. Н. (2002). «Асимметричные каталитические реакции Манниха, катализируемые производными мочевины: энантиоселективный синтез β-арил-β-аминокислот». Варенье. Chem. Soc. 124 (44): 12964–12965. Дои:10.1021 / ja028353g.
- ^ Yamamoto, H .; Момияма, Н. (сентябрь 2004 г.). «Кислотный катализ Бренстеда ахиральных енаминов для регио- и энантиоселективного синтеза нитрозоальдола». Варенье. Chem. Soc. 127 (4): 1080–1081. Дои:10.1021 / ja0444637. ЧВК 1460970. PMID 15669829.
- ^ Hine, J .; Linden, S.M .; Канагасабапати, В. М. (декабрь 1985 г.). «Катализ двойной водородной связи реакции фенилглицидилового эфира с диэтиламином под действием 1,8-бифенилендиола». J. Org. Chem. 50 (25): 5096–5099. Дои:10.1021 / jo00225a021.
- ^ Uyeda, C .; Якобсен, Э. Н. (июль 2008 г.). «Энантиоселективные перегруппировки Клейзена с катализатором-донором водородной связи». Варенье. Chem. Soc. 130 (29): 9228–9229. Дои:10.1021 / ja803370x. ЧВК 2547484. PMID 18576616.
- ^ Rawal, Viresh H .; Thadani, A.N .; Станкович, А. (2004). «Энантиоселективные реакции Дильса-Альдера, катализируемые водородными связями». PNAS. 101 (16): 5846–5850. Bibcode:2004ПНАС..101.5846Т. Дои:10.1073 / pnas.0308545101. ЧВК 395998. PMID 15069185.
- ^ Schmidtchen, F. P .; Бергер, М. (август 1997 г.). «Искусственные органические молекулы-хозяева для анионов». Chem. Rev. 97 (5): 1609–1646. Дои:10.1021 / cr9603845.
- ^ Raheem, I.T .; Thiara, P. S .; Петерсон, Э. А .; Якобсен, Э. Н. (август 2007 г.). "Энантиоселективная циклизация гидроксилактамов типа Пикте-Шпенглера: катализ доноров Н-связи путем связывания анионов". Варенье. Chem. Soc. 129 (44): 13404–13405. Дои:10.1021 / ja076179w. PMID 17941641.
- ^ Reisman, S.E .; Дойл, А. Г. (май 2008 г.). «Энантиоселективные добавки к ионам оксокарбения, катализируемые тиомочевиной». Варенье. Chem. Soc. 130 (23): 7198–7199. Дои:10.1021 / ja801514m. ЧВК 2574628. PMID 18479086.
- ^ Zuend, S.J .; Якобсен, Э. Н. (сентябрь 2009 г.). "Механизм энантиоселективного гидроцианирования имина, катализируемого амидо-тиомочевиной: стабилизация переходного состояния посредством множественных нековалентных взаимодействий". Варенье. Chem. Soc. 131 (42): 15358–15374. Дои:10.1021 / ja9058958. ЧВК 2783581. PMID 19778044.
- ^ Урагучи, Д .; Терада, М. (апрель 2004 г.). «Хиральные кислотные катализируемые Бренстедом прямые реакции Манниха посредством электрофильной активации». Варенье. Chem. Soc. 126 (17): 5356–5357. Дои:10.1021 / ja0491533. PMID 15113196.
- ^ Jansen, A.C.A .; Брюсси, Дж. (Май 1983 г.). «Высокостереоселективный синтез s (-) - [1,1'-бинафталин] -2,2'-диола». Tetrahedron Lett. 24 (31): 3261–3262. Дои:10.1016 / S0040-4039 (00) 88151-4.
- ^ Ли, А .; Стюарт, Дж. Д .; Clardy, J .; Ганем, Б. (апрель 1995 г.). «Новое понимание каталитического механизма хоризматмутаз из структурных исследований». Химия и биология. 2 (4): 195–203. Дои:10.1016/1074-5521(95)90269-4. PMID 9383421.
- ^ Ноулз, Р. Р .; Lin, S .; Якобсен, Э. Н. (апрель 2010 г.). «Энантиоселективная катионная полициклизация, катализируемая тиомочевиной». Варенье. Chem. Soc. 132 (14): 5030–5032. Дои:10.1021 / ja101256v. ЧВК 2989498. PMID 20369901.
- ^ Ван, Б .; Wu, F .; Wang, Y .; Лю, X .; Дэн Л. (январь 2007 г.). «Контроль диастереоселективности в тандемных асимметричных реакциях, генерирующих несмежные стереоцентры с бифункциональным катализом алкалоидами хинного дерева». Варенье. Chem. Soc. 129 (4): 768–769. Дои:10.1021 / ja0670409. PMID 17243806.
- ^ Веннемерс, Хельма (2011). «Асимметричный катализ пептидами». Chem. Сообщество. 47: 12036–12041. Дои:10.1039 / C1CC15237H.
- ^ Далко П. И. (2007). Энантиоселективный органокатализ. Вайнхайм: Wiley-VCH. ISBN 978-3-527-31522-2.
- ^ Сюй, Вэйчи; Ариено, Маркус; Лёв, Хенрик; Хуанг, Кайфан; Се, Сюлань; Крухтер, Томас; Ма, Цяо; Си Цзяньвэй; Хуанг, Бяо; Уист, Олаф; Гун, Лэй (20 июля 2016 г.). «Дизайн на основе металла: энантиоселективный катализ на водородных связях, требующий загрузки катализатора только частей на миллион». Журнал Американского химического общества. 138 (28): 8774–8780. Дои:10.1021 / jacs.6b02769. ISSN 0002-7863.
- ^ Шрайнер, Питер Р. (2003). «Безметалловый органокатализ посредством явных водородных связей». Chem. Soc. Rev. 32 (5): 289–296. Дои:10.1039 / B107298F. PMID 14518182.
- ^ Якобсен, Э. «Асимметричный катализ с хиральными донорами Н-связи» (PDF). Получено 18 декабря 2012.
- ^ Jacobsen, E. N .; Дастин, Дж. М .; Зуэнд, С. Дж. (Ноябрь 2008 г.). «Каталитический асимметричный тотальный синтез (+) - Йохимбина». Орг. Латыш. 10 (5): 745–748. Дои:10.1021 / ol702781q. PMID 18257582.
- ^ Такемото, Йошиджи; Миябе, Х. (июль 2008 г.). «Открытие и применение асимметричной реакции с помощью многофункциональных тиомочевин». Бык. Chem. Soc. JPN. 81 (7): 785–795. Дои:10.1246 / bcsj.81.785.
- ^ Zuend, S.J .; Coughlin, M. P .; Lalonde, M. P .; Якобсен, Э. Н. (октябрь 2009 г.). «Масштабируемый [sic] каталитический асимметричный синтез по Стрекеру неприродных альфа-аминокислот». Природа. 461 (7266): 968–970. Bibcode:2009Натура.461..968Z. Дои:10.1038 / природа08484. ЧВК 2778849. PMID 19829379.