Синдром Санджада-Сакати - Sanjad-Sakati syndrome
| Синдром Санджада-Сакати | |
|---|---|
| Другие имена | Синдром гипопаратиреоза, низкого роста, умственной отсталости |
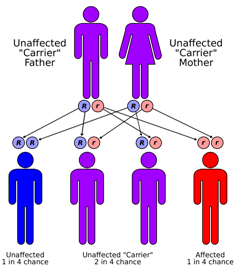 | |
| Синдром Санджада-Сакати наследуется по аутосомно-рецессивному типу. | |
| Обычное начало | диагноз ставится клинически по признакам триады задержки роста, дисморфических признаков и гипокальцемической тетании или приступов |
Синдром Санджада-Сакати редкий аутосомный рецессивное генетическое заболевание, наблюдаемое у потомков Ближневосточный происхождение. Впервые он был описан в Саудовской Аравии,[1] но его видели в Катаре, Кувейте, Омане и других детях с Ближнего Востока, а также из других мест.[2] Состояние вызвано мутациями или делециями в TBCE ген хромосомы №1.
Состояние характеризуется триадой задержки роста и Интеллектуальная недееспособность, гипопаратиреоз и дисморфизм.
Презентация
У детей с синдромом Санджада Сакати есть триада: а) гипопаратиреоз (с эпизодами гипокальциемия, гипокальциемический тетания и гипокальциемический припадки.b) тяжелая Интеллектуальная недееспособность иc) дисморфизм Обычно дети с этим синдромом рождаются с низкой массой тела из-за задержки внутриутробного развития. При рождении наблюдается дисморфизм, который позже типизируется в признаки, описанные ниже. Ребенок отстает в росте, часто с очевидными дефицит гормона роста и имеет умственную отсталость от умеренной до тяжелой, в основном в результате повторных судорог, вызванных низким уровнем ионного кальция в крови. Уровни иммуно-реактивного паратгормона от низкого до неопределяемого, с низким уровнем кальция и высоким уровнем фосфата в крови.[нужна цитата ]
Дисморфизм Наиболее ярко проявляется на лице со следующими чертами:[нужна цитата ]
- Длинное узкое лицо
- Глубоко посаженные маленькие глаза
- Клювый нос
- Большие гибкие уши
- Маленькая голова (микроцефалия ) и
- Тонкие губы с длинным желобком.
Другие преимущества
Другие функции включают:
- Задержка роста
- Маленькие руки и ноги с длинными сужающимися пальцами и клинодактилия
- Стоматологические аномалии в виде смещения и неправильный прикус
В другом исследовании с участием шести пациентов были выявлены низкие уровни IGF-1 и заметно замедленный костный возраст.[3]
Генетика
Это нарушение вызвано аномалией TBCE ген,[4] локус для которого находится на хромосоме 1q42.3. Локус представляет собой область гена размером 230 т.п.н. с идентифицированными делециями и мутациями у пораженных людей.[5] В редких случаях заболевание не связано с аномалией гена TBCE.[6]
Диагностика
Этот раздел пуст. Вы можете помочь добавляя к этому. (Август 2017 г.) |
Управление
Лечение в основном является поддерживающим, контролируя судороги и уровень кальция в крови.[нужна цитата ]
История
Первое сообщение из Саудовской Аравии в 1988 г. Синдром Санджада-Сакати,[7] также известен как Синдром гипопаратиреоза-ретардации-дисморфизма (HRD), или реже как Ближневосточный синдром, это очень редкое генетически наследуемое заболевание, наблюдаемое на Ближнем Востоке и у детей ближневосточного происхождения в других странах мира. Состояние названо в честь Сами А. Санджад и Надя Авни Сакати.[нужна цитата ]
использованная литература
- ^ Санджад, S (1988). «Врожденный гипопаратиреоз с дисморфическими признаками: новый синдром. (Аннотация)». Педиатрические исследования. 23: 271A.
- ^ Sanjad, S.A .; Сакати, Н. А .; Абу-Осба, Ю.К .; Kaddoura, R .; Милнер, Р. Д. (февраль 1991 г.). «Новый синдром врожденного гипопаратиреоза, тяжелой недостаточности роста и дисморфических особенностей». Архив детских болезней. 66 (2): 193–196. Дои:10.1136 / adc.66.2.193. ISSN 1468-2044. ЧВК 1792808. PMID 2001103.
- ^ Hershkovitz, E .; Шалитин, С .; Levy, J .; Leiberman, E .; Weinshtock, A .; Varsano, I .; Городищер Р. (май 1995 г.). «Новый синдром врожденного гипопаратиреоза, связанный с дисморфизмом, задержкой роста и задержкой развития - отчет шести пациентов». Израильский журнал медицинских наук. 31 (5): 293–297. ISSN 0021-2180. PMID 7538982.
- ^ «Запись OMIM - * 604934 - Тубулин-специфический шаперон E; TBCE». Omim.org. Получено 2015-08-25.
- ^ Парвари, Р., Гершковиц, Э., Гроссман, Н., Городишер, Р., Лойс, Б., Зечич, А., Мортье, Г., Грегори, С., Шарони, Р., Камбурис, М., Сакати, Н., Мейер, Б.Ф. и 10 других. Мутация TBCE вызывает гипопаратиреоз-задержку-дисморфизм и аутосомно-рецессивный синдром Кенни-Каффи. Nature Genet. 32: 448-452, 2002.
- ^ Courtens, W., Wuyts, W., Poot, M., Szuhai, K., Wauters, J., Reyniers, E., Eleveld, M., Diaz, G., Nothen, MM, Parvari, R., Hypoparathyroidism- синдром ретардации-дисморфизма у девочки: новый вариант, не вызванный мутацией TBCE - клинический отчет и обзор. Am. J. Med. Genet. 140A: 611-617, 2006.
- ^ "Запись в OMIM - № 241410 - Синдром гипопаратиреоза, задержки развития и дисморфизма; HRD". Omim.org. Получено 2015-08-25.
внешние ссылки
| Классификация | |
|---|---|
| Внешние ресурсы |