Сравнение термодинамического и кинетического контроля реакции - Thermodynamic versus kinetic reaction control - Wikipedia

Термодинамический контроль реакции или же управление кинетической реакцией в химическая реакция может определять состав смеси продуктов реакции, когда конкурирующие пути приводят к разным продуктам, а условия реакции влияют на избирательность или же стереоселективность. Это различие актуально, когда продукт А формируется быстрее, чем продукт B поскольку энергия активации для продукта А ниже, чем для продукта B, но продукт B более стабильный. В таком случае А является кинетическим продуктом и предпочтительнее при кинетическом контроле и B является термодинамическим продуктом и предпочтительнее при термодинамическом контроле.[1][2][3]
Условия реакции, такие как температура, давление или растворитель, влияют на то, какой путь реакции может быть предпочтительным: либо кинетически контролируемый, либо термодинамически контролируемый. Обратите внимание, это верно только в том случае, если энергия активации двух путей различается, причем один путь имеет более низкую Eа (энергия активации ), чем другой.
Преобладание термодинамического или кинетического контроля определяет конечный состав продукта, когда эти конкурирующие пути реакции приводят к различным продуктам. Условия реакции, как указано выше, влияют на избирательность реакции - т.е. какой путь выбран.
Асимметричный синтез Это область, в которой особенно важно различать кинетический и термодинамический контроль. Поскольку пары энантиомеров для всех целей и задач имеют одинаковую свободную энергию Гиббса, термодинамический контроль приведет к рацемическая смесь по необходимости. Таким образом, любой каталитический реакция, которая дает продукт с ненулевым энантиомерный избыток находится под хотя бы частичным кинетическим контролем. (Во многих стехиометрический При асимметричных превращениях энантиомерные продукты фактически образуются в виде комплекса с источником хиральности перед стадией обработки реакции, что технически делает реакцию диастереоселективной. Хотя такие реакции все еще обычно контролируются кинетически, термодинамическое регулирование в принципе возможно.)
Объем
В реакциях Дильса – Альдера
В Реакция Дильса – Альдера из циклопентадиен с фуран может произвести два изомерный товары. В комнатная температура, преобладает кинетический контроль реакции и менее стабильный эндо-изомер 2 является основным продуктом реакции. При 81 ° C и после длительного времени реакции химическое равновесие может заявить о себе и термодинамически более стабильный экзо-изомер 1 сформирован.[4] В экзо продукт более стабилен за счет более низкой степени стерическая гиперемия, в то время как эндо продукту способствует перекрытие орбит в переходное состояние.

Выдающийся и очень редкий пример полный кинетический и термодинамический контроль реакции в процессе тандем меж- / внутримолекулярная реакция Дильса – Альдера бис-фурилдиенов 3 с гексафтор-2-бутин или же диметилацетилендикарбоксилат (DMAD) были обнаружены и описаны в 2018 году.[5][6] При низкой температуре протекают реакции хемоселективно приводящие исключительно к аддуктам клещевого - [4 + 2] циклоприсоединения (5). Эксклюзивное образование домино -аддукты (6) наблюдается при повышенных температурах.
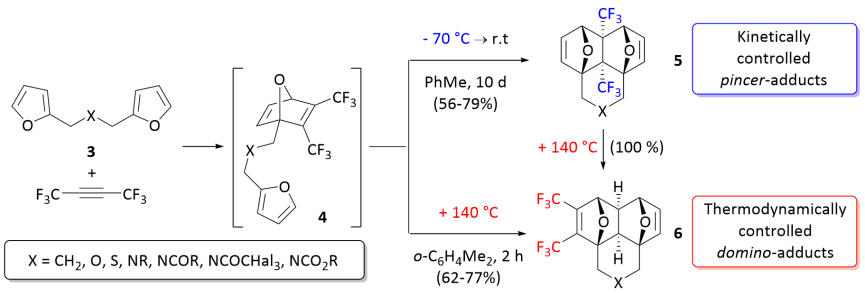
Теоретические расчеты DFT реакции между гексафтор-2-бутин и диены 3а-c были выполнены. Реакция, начинающаяся с [4 + 2] циклоприсоединения CF3C≡CCF3 в одном из фурановых фрагментов происходит согласованным образом через TS1 и представляет собой этап ограничения скорости всего процесса с активационный барьер Δграмм‡ ≈ 23,1–26,8 ккал / моль.
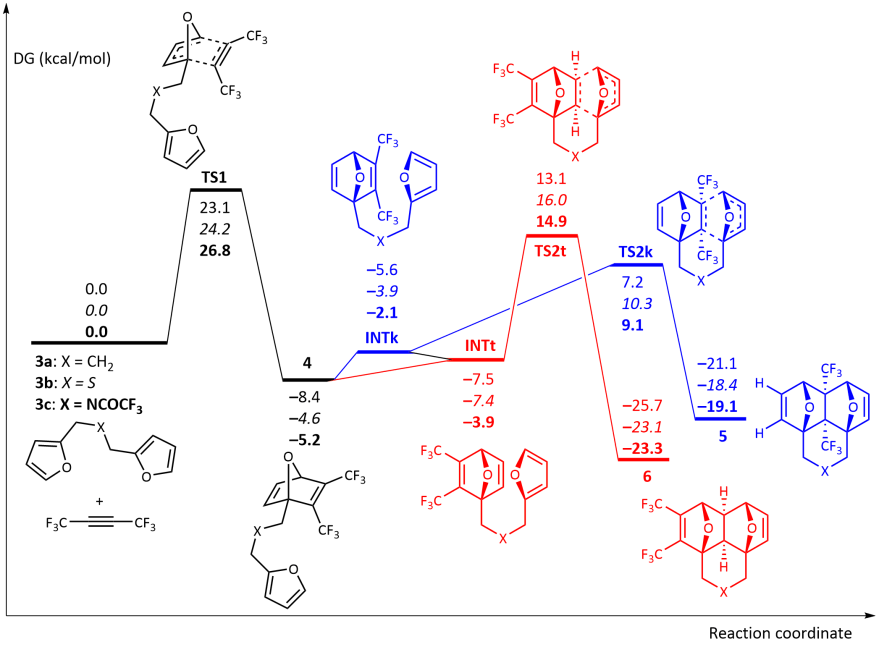
Далее реакция могла продолжаться через два конкурирующих канала, т.е. либо ведущие к продуктам типа клещей 5 через TS2k или приводящий к образованию продукта домино 6 через TS2t. Расчеты показали, что кинетически более выгоден первый канал (Δграмм‡ ≈ 5,7–5,9 ккал / моль). Между тем, изделия домино 6 более термодинамически стабильны, чем 5 (Δграмм‡ ≈ 4,2-4,7 ккал / моль), что может вызвать изомеризацию 5 в 6 при повышенной температуре. Действительно, рассчитанные активационные барьеры для 5 → 6 изомеризация через ретро-реакция Дильса – Альдера 5 с последующим внутримолекулярным [4 + 2] -циклоприсоединением в промежуточной цепи 4 давать 6 составляют 34,0–34,4 ккал / моль.
В енолятной химии
в протонирование из енолят-ион кинетическим продуктом является энол а термодинамический продукт представляет собой кетон или же альдегид. Карбонильные соединения и их энолы быстро меняются протон переводы, катализируемые кислоты или же базы, даже в следовых количествах, в этом случае опосредовано енолятом или источником протонов.
в депротонирование несимметричного кетон кинетическим продуктом является энолировать в результате удаления наиболее доступного α-H, в то время как термодинамический продукт имеет более сильно замещенный енолятный фрагмент.[7][8][9][10] Использование низких температур и стерильности базы увеличивает кинетическую селективность. Здесь разница в пKб между основанием и енолятом настолько велико, что реакция по существу необратима, поэтому уравновешивание, приводящее к термодинамическому продукту, вероятно, является протонным обменом, происходящим во время добавления между кинетическим енолятом и еще непрореагировавшим кетоном. Обратное добавление (добавление кетона к основанию) с быстрым перемешиванием минимизирует это. Положение равновесия будет зависеть от противокатиона и растворителя.

Если использовать гораздо более слабое основание, депротонирование будет неполным, и будет равновесие между реагентами и продуктами. Термодинамический контроль достигается, однако реакция остается неполной, если енолят продукта не улавливается, как в приведенном ниже примере. Поскольку перенос H происходит очень быстро, а реакция захвата протекает медленнее, соотношение захваченных продуктов в значительной степени отражает равновесие депротонирования.

В электрофильных добавках
В электрофильная добавка реакция бромистый водород к 1,3-бутадиен температура выше комнатной приводит преимущественно к термодинамически более стабильному 1,4-аддукту, 1-бром-2-бутену, но снижение температуры реакции до температуры ниже комнатной способствует кинетическому 1,2-аддукту, 3-бром-1-бутену.[3]

- Причина разной селективности заключается в следующем: Оба продукта являются результатом Марковников протонирование в положении 1, в результате чего резонанс -стабилизированный аллильный катион. 1,4-аддукт помещает больший атом Br в менее загруженный сайт и включает более сильно замещенный алкеновый фрагмент, в то время как 1,2-аддукт является результатом атаки нуклеофила (Br−) на углерод аллильного катиона, несущего наибольший положительный заряд (более высокозамещенный углерод является наиболее вероятным местом для положительного заряда).

Характеристики
- В принципе, каждая реакция находится на континууме между чистым кинетическим контролем и чистым термодинамическим контролем. Эти термины относятся к данной температуре и шкале времени. Процесс приближается к чисто кинетическому управлению при низкой температуре и коротком времени реакции. В течение достаточно длительного времени каждая реакция приближается к чистому термодинамическому контролю, по крайней мере, в принципе. Этот временной масштаб становится короче при повышении температуры.
- В каждой реакции первым образуется продукт, который легче всего образуется. Таким образом, каждая реакция априори запускается под кинетическим контролем.[11]
- Необходимым условием термодинамического контроля является обратимость или механизм, позволяющий уравновешивать продукты. Считается, что реакции происходят под термодинамическим контролем реакции, когда обратная реакция протекает достаточно быстро, чтобы равновесие устанавливает себя в течение отведенного времени реакции. Таким образом, всегда предпочтение отдается термодинамически более стабильному продукту.
- При кинетическом контроле реакции одна или обе прямые реакции, приводящие к возможным продуктам, проходят значительно быстрее, чем уравновешивание между продуктами. По истечении времени реакции т, соотношение продуктов - это отношение констант скорости k и, таким образом, функция разницы энергий активации Eа или Δграмм‡:
- (уравнение 1)
![ln left ({ frac {[A] _ {t}} {[B] _ {t}}} right) = ln left ({ frac {k_ {A}} {k_ {B}) }} right) = - { frac { Delta E_ {a}} {RT}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/d3c4abce8554f016a84af1e7dad83761b5dd71d7)
- Если не предотвратить уравновешивание (например, путем удаления продукта из реакционной смеси, как только он образуется), «чистый» кинетический контроль, строго говоря, невозможен, потому что некоторое количество уравновешивания будет иметь место до того, как реагенты полностью израсходуются. На практике многие системы хорошо аппроксимируются как работающие с кинетическим управлением из-за пренебрежимо медленного уравновешивания. Например, многие энантиоселективные каталитические системы обеспечивают почти энантиочистый продукт (> 99% ее), даже несмотря на то, что энантиомерные продукты имеют такую же свободную энергию Гиббса и одинаково предпочтительны с термодинамической точки зрения.
- При чисто термодинамическом управлении реакцией, когда равновесие достигнуто, распределение продукта будет функцией стабильности грамм°. После бесконечного времени реакции соотношение концентраций продукта будет равно константа равновесия Kэкв и, следовательно, быть функцией разницы в Свободные энергии Гиббса,
- (уравнение 2)
- В принципе, «чистый» термодинамический контроль также невозможен, поскольку равновесие достигается только после бесконечного времени реакции. На практике, если А и B преобразовать с общими константами скорости kж и kр, то для большинства практических целей изменение состава становится незначительным после т ~ 3.5/(kж + kр), или приблизительно пять периодов полураспада, и соотношение продуктов системы можно рассматривать как результат термодинамического контроля.
![ln left ({ frac {[A] _ {{ infty}}} {[B] _ {{ infty}}}} right) = ln K _ {{eq}} = - { гидроразрыв { Delta G ^ { circ}} {RT}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/fe18ae931f200f667234f9ff8c2033ba94c98205)
- Как правило, короткое время реакции способствует кинетическому контролю, тогда как более длительное время реакции способствует термодинамическому контролю реакции. Низкие температуры улучшают селективность в обоих наборах условий, поскольку Т находится в знаменателе в обоих случаях. Идеальной температурой для оптимизации выхода наиболее быстро образующегося продукта будет самая низкая температура, которая обеспечит завершение реакции за разумный промежуток времени.[12] Идеальная температура для реакции под термодинамическим контролем - это самая низкая температура, при которой равновесие будет достигнуто за разумное время.[13] При необходимости селективность можно повысить, затем медленно охлаждая реакционную смесь, чтобы еще больше сдвинуть равновесие в сторону наиболее стабильного продукта. Когда разница в стабильности продукта очень велика, термодинамически контролируемый продукт может доминировать даже в относительно жестких условиях реакции.
- Если реакция находится под термодинамическим контролем при данной температуре, она также будет находиться под термодинамическим контролем при более высокой температуре в течение того же времени реакции.
- Таким же образом, если реакция находится под кинетическим контролем при данной температуре, она также будет находиться под кинетическим контролем при любой более низкой температуре в течение того же времени реакции.
- Если предположить, что будет новая реакция априори при кинетическом контроле можно обнаружить наличие механизма уравновешивания (и, следовательно, возможность термодинамического контроля), если распределение продукта:
- меняется со временем,
- показывает, что один продукт доминирует при одной температуре, а другой доминирует при другой температуре (инверсия доминирования), или
- изменяется с температурой, но не согласуется с уравнением 1, то есть изменение температуры (без изменения времени реакции) вызывает изменение соотношения продуктов которая больше или меньше, чем можно было бы ожидать только по изменению температуры, при условии, что практически не зависит от температуры в умеренном температурном диапазоне.[14]
- Таким же образом можно обнаружить возможность кинетического управления, если изменение температуры вызывает изменение соотношения продуктов, которое несовместимо с уравнением 2, при условии, что практически не зависит от температуры в умеренном температурном диапазоне.[15]
![{[A] _ {t}} / {[B] _ {t}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/6e66460d660f8572d9b47c3943b17e49c35a4048)


История
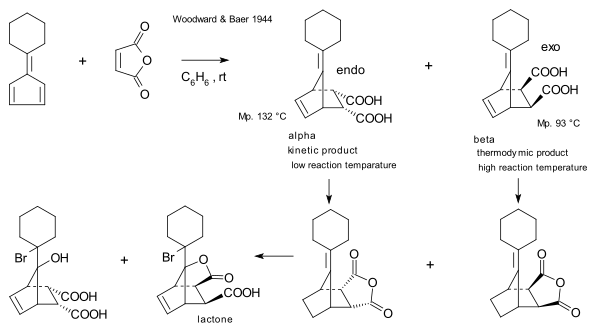
Первыми, кто сообщил о взаимосвязи между кинетическим и термодинамическим контролем, были Р. Б. Вудворд и Гарольд Баер в 1944 году.[16] Они повторно исследовали реакцию между малеиновый ангидрид и Fulvene впервые сообщил в 1929 г. Отто Дильс и Курт Алдер.[17] Они отметили, что в то время как эндо-изомер образуется быстрее, более длительное время реакции, а также относительно повышенные температуры приводят к более высоким отношениям экзо / эндо который должен был быть рассматривается в свете замечательной стабильности экзо-соединения, с одной стороны, и очень легкой диссоциации эндо-изомера, с другой.
К. К. Ингольд с Э. Д. Хьюз и Г. Кэтчпол независимо описал термодинамическую и кинетическую модель управления реакцией в 1948 году.[18] Они повторно исследовали определенные аллильная перегруппировка сообщенный в 1930 г. Якоб Мейсенхаймер.[19] Сольволиз гамма-фенилаллилхлорида с AcOK в уксусной кислоте, как было обнаружено, дает смесь гамма- и альфа-ацетата, причем последний превращается в первый при уравновешивании. Это было интерпретировано как случай в области анионотропии явления, известного по прототропии, различия между кинетическим и термодинамическим контролем в ионной рекомбинации.

Рекомендации
- ^ Органическая химия, 3-е изд., М. А. Фокс и Дж. К. Уайтселл, Джонс и Бартлетт, 2004 г. ISBN 0-7637-2197-2
- ^ Руководство по механизму в органической химии, 6-е издание, Питер Сайкс, Pearson Prentice Hall, 1986. ISBN 0-582-44695-3
- ^ а б Введение в органическую химию I, Сет Роберт Эльсхаймер, Blackwell Publishing, 2000 ISBN 0-632-04417-9
- ^ Продвинутая органическая химия, часть A: структура и механизмы, 5-е изд., Фрэнсис А. Кэри, Ричард Дж. Сандберг, 2007 г. ISBN 978-0-387-44899-2
- ^ Ксения К. Борисова, Елизавета А. Квятковская, Евгения В. Никитина, Ринат Р. Айсин, Роман А. Новиков, Федор И. Зубков. «Классический пример полного кинетического и термодинамического контроля. Реакция Дильса-Альдера между DMAD и бис-фурилдиенами ». J. Org. Chem., 2018, 83 (8), стр 4840-4850. DOI: 10.1021 / acs.joc.8b00336 https://pubs.acs.org/doi/abs/10.1021/acs.joc.8b00336
- ^ Ксения К. Борисова, Евгения В. Никитина, Роман А. Новиков, Виктор Н. Хрусталев, Павел В. Дороватовский, Ян В. Зубавичус, Максим Леонидович Кузнецов, Владимир П. Зайцев, Алексей В. Варламов и Федор И. Зубков. «Реакции Дильса-Альдера между гексафтор-2-бутином и бис-фурилдиенами: кинетический и термодинамический контроль». Chem. Commun., 2018, 54, с. 2850-2853. DOI: 10.1039 / c7cc09466c http://pubs.rsc.org/en/content/articlelanding/2018/cc/c7cc09466c#!divAbstract
- ^ Термодинамический продукт против кинетического продукта
- ^ Жан д'Анджело, отчет Tetrahedron номер 25: Кетоновые енолаты: региоспецифические препараты и синтетические применения, Тетраэдр, Том 32, Выпуск 24, 1976 г., страницы 2979-2990, ISSN 0040-4020, Дои:10.1016/0040-4020(76)80156-1
- ^ Химия карбанионов. IX. Енолиты калия и лития, полученные из циклических кетонов Герберт О. Хаус, Барри М. Трост J. Org. Chem., 1965, 30 (5), стр. 1341–1348 Дои:10.1021 / jo01016a001
- ^ Химия карбанионов. XV. Стереохимия алкилирования 4-трет-бутилциклогексанона Герберт О. Хаус, Бен А. Тефертиллер, Хью Д. ОлмстедJ. Орг. Chem., 1968, 33 (3), стр. 935–942 Дои:10.1021 / jo01267a002
- ^ Это неверно, только если последующее уравновешивание происходит так же быстро или быстрее.
- ^ Если не довольствоваться незавершенной реакцией, тогда может потребоваться отделение продукта от непрореагировавшего исходного материала.
- ^ В худшем случае, Kэкв приблизится к 1 как Т повышается, и доля наиболее стабильного продукта будет стремиться к 50% реакционной смеси.
- ^ будет не зависеть от температуры или почти не зависит, если мала, что было бы так, если бы этапы определения нормы, ведущие к каждому продукту, были одинаковыми молекулярность, например, если оба столкнулись с одним и тем же реагентом.
- ^ будет не зависеть от температуры или почти не зависит, если мала, что было бы так, если бы общие преобразования для каждого продукта были одинаковыми молекулярность, например, если бы обе были фрагментацией молекулы с образованием пары молекул или если бы обе были конденсацией двух молекул с образованием единой молекулы.
- ^ Исследования реакций присоединения диенов. II.1 Реакция 6,6-пентаметиленфульвена с малеиновым ангидридом Р. Б. Вудворд, Гарольд Бэр Дж. Ам. Chem. Soc., 1944, 66 (4), стр. 645–649. Дои:10.1021 / ja01232a042
- ^ Дильс, О. и Алдер, К. (1929), Synthesen in der hydroaromatischen Reihe, IV. Mitteilung: Über die Anlagerung von Maleinsäure-anhydrid an arylierte Diene, Triene und Fulvene (Mitbearbeitet von Paul Pries). Berichte der deutschen chemischen Gesellschaft (серии A и B), 62: 2081–2087. Дои:10.1002 / cber.19290620829
- ^ Перегруппировка и замещение в анионотропных системах. Часть III. Механизм и равновесие анионотропного изменения A. G. Catchpole, E. D. Hughes и C. K. Ingold J. Chem. Soc., 1948, 8-17 Дои:10.1039 / JR9480000008
- ^ Мейсенхаймер Дж. И Линк Дж. (1930), Über die Verschiebung in der Allyl-Gruppe. 3. Mitteilung über Замена и добавление. Юстус Либигс Аннален дер Хеми, 479: 211–277. Дои:10.1002 / jlac.19304790114

