Резонанс (химия) - Resonance (chemistry)

В химия, резонанс это способ описания связи в определенных молекулах или ионах комбинацией нескольких содействующие структуры (или же формы,[1] также известный как резонансные структуры или же канонические структуры) в резонансный гибрид (или же гибридная структура) в теория валентной связи. Он имеет особое значение для описания делокализованные электроны в пределах определенных молекулы или же многоатомные ионы где связь не может быть выражена одним единственным Структура Льюиса.
Обзор
В рамках теория валентной связи, резонанс - это расширение идеи о том, что связь в химические вещества можно описать структурой Льюиса. Для многих химических соединений одной структуры Льюиса, состоящей из атомов, подчиняющихся правилу октетов, возможно несущих формальные заряды и связанных связями положительного целого порядка, достаточно для описания химической связи и рационализации экспериментально определенных молекулярных свойств, таких как длины связей, углы , и дипольный момент.[2] Однако в некоторых случаях можно было нарисовать более одной структуры Льюиса, и экспериментальные свойства несовместимы с какой-либо одной структурой. Чтобы справиться с ситуацией такого типа, несколько структур рассматриваются вместе как среднее значение, и говорят, что молекула представлена резонансным гибридом, в котором несколько структур Льюиса используются вместе для описания ее истинной структуры.
Например, в NO2–, нитрит аниона, длины двух связей N – O равны, даже если ни одна структура Льюиса не имеет двух связей N – O с одинаковыми формальными ордер на облигации. Однако его измеренная структура согласуется с описанием как резонансного гибрида двух основных структур, показанных выше: он имеет два равный Связи N – O 125 мкм, промежуточные по длине между типичной одинарной связью N – O (145 мкм в гидроксиламин, H2N – OH) и двойной связи N – O (115 пм в ион нитрония, [O = N = O]+). В соответствии с соответствующими структурами каждая связь N – O представляет собой в среднем формальную одинарную и формальную двойную связь, что приводит к истинному порядку связи 1,5. Благодаря этому усреднению, описание Льюиса связи в NO2– согласуется с экспериментальным фактом, что анион имеет эквивалентные связи N – O.
Резонансный гибрид представляет реальную молекулу как "среднее" из структур, способствующих, с длинами связей и частичные сборы принимая промежуточные значения по сравнению с ожидаемыми для отдельных структур Льюиса вкладчиков, если бы они существовали как «настоящие» химические образования.[3] Способствующие структуры различаются только формальный распределение электронов по атомам, а не в реальной физически и химически значимой электронной или спиновой плотности. Хотя структуры вкладчиков могут отличаться формальными поручениями на облигации и официальное обвинение задания, все участвующие структуры должны иметь одинаковое количество валентных электронов и одинаковый спин множественность.[4]
Поскольку делокализация электронов снижает потенциальную энергию системы, любой вид, представленный резонансным гибридом, более стабилен, чем любая из (гипотетических) структур, вносящих свой вклад.[5] Разница в потенциальной энергии между фактическими видами и (вычисленной) энергией вносящей вклад структуры с самой низкой потенциальной энергией называется резонансная энергия[6] или энергия делокализации. Величина резонансной энергии зависит от предположений, сделанных относительно гипотетических «нестабилизированных» частиц и используемых вычислительных методов, и не представляет собой измеримую физическую величину, хотя сравнения резонансных энергий, вычисленных при аналогичных предположениях и условиях, могут иметь химический смысл.
Молекулы с расширенной π-системой, такие как линейные полиены и полиароматические соединения, хорошо описываются резонансными гибридами, а также делокализованными орбиталями в теория молекулярных орбиталей.
Резонанс против изомерии
Резонанс следует отличать от изомерия. Изомеры представляют собой молекулы с одинаковой химической формулой, но представляют собой разные химические соединения с различным расположением атомных ядер в пространстве. С другой стороны, компоненты резонанса в молекуле могут различаться только способом, которым электроны формально приписываются атомам в структуре Льюиса. изображения молекулы. В частности, когда говорят, что молекулярная структура представлена резонансным гибридом, это нет означают, что электроны молекулы «резонируют» или перемещаются вперед и назад между несколькими наборами положений, каждое из которых представлено структурой Льюиса. Скорее это означает, что набор способствующих структур представляет собой промежуточную структуру (средневзвешенное значение участников) с единственной четко определенной геометрией и распределением электронов. Неверно рассматривать резонансные гибриды как быстро взаимопревращающие изомеры, даже если термин «резонанс» может вызвать такое представление.[7] (Как описано ниже термин "резонанс" возник как аналогия классической физики квантово-механического явления, поэтому его не следует толковать слишком буквально.) Символично, двунаправленная стрелка используется для обозначения того, что A и B вносят вклад в формы одного химического вещества (в отличие от стрелки равновесия, например, ; видеть ниже для подробностей по использованию).


Показательна нехимическая аналогия: можно описать характеристики реального животного, нарвал, с точки зрения характеристик двух мифических существ: единорог, существо с единственным рогом на голове, и левиафан, большое, похожее на кита существо. Нарвал - это не существо, которое колеблется между единорогом и левиафаном, а единорог и левиафан не имеют физического существования вне коллективного человеческого воображения. Тем не менее, описание нарвала в терминах этих воображаемых существ дает достаточно хорошее описание его физических характеристик.
Из-за путаницы с физическим значением слова резонанс, поскольку на самом деле никакие объекты физически не "резонируют", было предложено отказаться от термина "резонанс" в пользу делокализация[8] и резонансной энергии отказались в пользу энергия делокализации. Резонансная структура становится способствующая структура и резонансный гибрид становится гибридная структура. Двуглавые стрелки будут заменены запятыми, чтобы проиллюстрировать набор структур, поскольку стрелки любого типа могут указывать начинающим студентам на то, что происходит химическое изменение.
Представление в схемах
![{ Displaystyle { ce {[S = C = N ^ { ominus} <-> ^ { ominus} ! S-C { Equiv} N]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/cc41b6c9b3f1ef739335969a581e9a66d5ed0b1a)

На диаграммах участвующие структуры обычно разделяются двуглавыми стрелками (↔). Стрелку не следует путать с направлением вправо и влево. стрелка равновесия (⇌). Все структуры вместе могут быть заключены в большие квадратные скобки, чтобы указать, что они представляют собой одну единственную молекулу или ион, а не разные виды в одном химическое равновесие.
В качестве альтернативы использованию вспомогательных структур в диаграммах может использоваться гибридная структура. В гибридной структуре пи-облигации которые участвуют в резонансе, обычно изображаются кривыми[9] или пунктирные линии, указывающие, что это частичные, а не нормальные полные пи-связи. В бензоле и других ароматических кольцах делокализованные пи-электроны иногда изображаются в виде сплошного круга.[10]
История
Концепция впервые появилась в 1899 году в Йоханнес Тиле "Гипотеза частичной валентности" для объяснения необычной стабильности бензола, которой нельзя было ожидать от Август Кекуле Структура России предложена в 1865 году с чередованием одинарных и двойных связей.[11] Бензол претерпевает реакции замещения, а не реакции присоединения, что типично для алкены. Он предположил, что связь углерод-углерод в бензоле является промежуточной между одинарной и двойной связью.
Предложение резонанса также помогло объяснить количество изомеров производных бензола. Например, структура Кекуле предсказывала четыре изомеры дибромбензола, в том числе два орто изомеры с бромированными атомами углерода, соединенными одинарной или двойной связью. В действительности существует только три изомера дибромбензола, и только один является орто, что согласуется с идеей, что существует только один тип углерод-углеродной связи, промежуточный между одинарной и двойной связью.[12]
Механизм резонанса был введен в квантовая механика к Вернер Гейзенберг в 1926 г. при обсуждении квантовых состояний атома гелия. Он сравнил структуру атома гелия с классической системой резонансных связанных гармонические осцилляторы.[3][13] В классической системе связь дает две моды, одна из которых ниже частота чем любая из несвязанных колебаний; квантово-механически эта более низкая частота интерпретируется как более низкая энергия. Линус Полинг использовал этот механизм для объяснения парциальной валентности молекул в 1928 году и развил его в серии статей в 1931-1933 годах.[14][15] Альтернативный термин мезомерия[16] популярный в немецких и французских публикациях с тем же значением был представлен К. К. Ингольд в 1938 г., но не получил широкого распространения в английской литературе. Текущая концепция мезомерный эффект приобрело родственное, но иное значение. Двунаправленная стрелка была введена немецким химиком. Фриц Арндт кто предпочел немецкую фразу Zwischenstufe или же промежуточный этап.
В Советском Союзе теория резонанса, особенно разработанная Полингом, подверглась критике в начале 1950-х годов как противоречащая марксистским принципам диалектический материализм, а в июне 1951 г. - Академия наук СССР под руководством Александр Несмеянов созвал конференцию по химическому строению органических соединений, в которой приняли участие 400 физиков, химиков и философов, на которой " псевдонаучный раскрыта и разоблачена суть теории резонанса ».[17]
Основные и второстепенные участники
Одна вносящая вклад структура может больше напоминать реальную молекулу, чем другая (в смысле энергии и стабильности). Конструкции с низким значением потенциальной энергии более устойчивы, чем конструкции с высокими значениями, и больше напоминают реальную структуру. Наиболее стабильные способствующие структуры называются основные участники. Энергетически невыгодные и, следовательно, менее благоприятные конструкции второстепенные участники. С правилами, перечисленными в приблизительном порядке убывания важности, основными участниками обычно являются структуры, которые
- как можно больше подчиняться Правило октета (8 валентных электронов вокруг каждого атома вместо недостатка или избытка, или 2 электрона для Элементы периода 1 );
- иметь максимальное количество ковалентных связей;
- нести минимум формально заряженных атомов, с минимальным и максимальным разделением разноименных и одинаковых зарядов соответственно;
- поместите отрицательный заряд, если он есть, на самый электроотрицательный атомы и положительный заряд, если есть, на самом электроположительном;
- не отклоняются существенно от идеализированных длин и углов связи (например, относительная незначительность вкладов резонанса типа Дьюара для бензола);
- поддерживать ароматические субструктуры локально, избегая антиароматических (видеть Клар секстет и бифенилен ).
Максимум восемь валентных электронов строго для Период 2 элемента Be, B, C, N, O и F, как максимум два для H и He, а также эффективно для Li.[18] Вопрос о расширении валентной оболочки третьего периода и более тяжелых элементов основной группы является дискуссионным. Структура Льюиса, в которой центральный атом имеет количество валентных электронов больше восьми, традиционно предполагает участие d-орбиталей в связывании. Однако общее мнение заключается в том, что, хотя они могут вносить незначительный вклад, участие d-орбиталей не имеет значения, и связь так называемых гипервалентный молекулы, по большей части, лучше объясняются вносящими вклад формами с разделенными зарядами, которые изображают трехцентровая четырехэлектронная связь. Тем не менее, по традиции, расширенные структуры октетов все еще обычно используются для таких функциональных групп, как сульфоксиды, сульфоны, и илиды фосфора, Например. Рассматриваемый как формализм, который не обязательно отражает истинную электронную структуру, IUPAC предпочитает такие изображения структурам с частичными связями, разделением зарядов или дательными связями.[19]
Эквивалентные участники вносят равный вклад в реальную структуру, в то время как важность неэквивалентных участников определяется степенью их соответствия свойствам, перечисленным выше. Большее количество структур, вносящих значительный вклад, и более объемное пространство, доступное для делокализованных электронов, приводят к стабилизации (снижению энергии) молекулы.
Примеры
Ароматические молекулы
В бензол два циклогексатриена Кекуле конструкции, впервые предложенные Кекуле, взяты вместе как составляющие структуры для представления общей структуры. В гибридной структуре справа пунктирный шестиугольник заменяет три двойные связи и представляет шесть электронов в наборе из трех молекулярные орбитали из π симметрия, с узловая плоскость в плоскости молекулы.
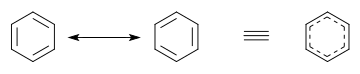
В фуран а одинокая пара атома кислорода взаимодействует с π-орбиталями атомов углерода. В изогнутые стрелки изобразить перестановку делокализованные π электроны, что приводит к разным участникам.

Молекулы, богатые электронами
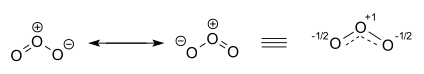
В озон Молекула представлена двумя участвующими структурами. На самом деле два концевых атома кислорода эквивалентны, а гибридная структура изображена справа с зарядом -1⁄2 на атомах кислорода и частичных двойных связях сплошной и штриховой линией и ордер на облигации 1 1⁄2.[20][21]
За гипервалентные молекулы, описанная выше рационализация может быть применена для создания структур, способствующих объяснению связывания в таких молекулах. Ниже показаны вспомогательные структуры 3c-4e связь в дифторид ксенона.
![{ Displaystyle { ce {[{ mathsf {F-XeF ^ {-} <-> F ^ {-} Xe-F}}]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/6841f8221ad24ec2c691c3280284c43996ad1ffb)
Электронно-дефицитные молекулы
В аллильный катион имеет две составляющие структуры с положительным зарядом на концевых атомах углерода. В гибридной структуре их заряд +1⁄2. Полный положительный заряд также может быть изображен как делокализованный между тремя атомами углерода.

В диборан Молекула описывается структурами, каждая из которых имеет дефицит электронов на разных атомах. Это уменьшает дефицит электронов на каждом атоме и стабилизирует молекулу. Ниже представлены структуры, способствующие индивидуальному 3c-2e связь в диборане.
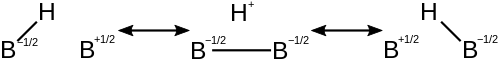
Реактивные промежуточные продукты
Часто реактивные промежуточные соединения, такие как карбокатионы и свободные радикалы имеют более делокализованную структуру, чем их исходные реагенты, что приводит к неожиданным продуктам. Классический пример: аллильная перегруппировка. Когда 1 моль HCl добавляется к 1 моль 1,3-бутадиена, в дополнение к обычно ожидаемому продукту 3-хлор-1-бутену мы также обнаруживаем 1-хлор-2-бутен. Эксперименты по изотопному мечению показали, что здесь происходит то, что дополнительная двойная связь смещается из положения 1,2 в положение 2,3 в некоторых частях продукта. Это и другие доказательства (например, ЯМР в суперкислотный растворов) показывает, что промежуточный карбокатион должен иметь сильно делокализованную структуру, отличную от его в основном классической (делокализация существует, но является небольшой) родительской молекулой. Этот катион (аллильный катион) можно представить с помощью резонанса, как показано выше.
Это наблюдение большей делокализации в менее стабильных молекулах является довольно общим. Возбужденные состояния сопряженных диены стабилизируются больше за счет конъюгации, чем их основные состояния, в результате чего они становятся органическими красителями.
Хорошо изученный пример делокализации без π-электронов (сверхсопряжение ) можно наблюдать в неклассических 2-норборнил катион. Другой пример метаний (CH+
5). Их можно рассматривать как содержащие трехцентровые двухэлектронные связи и представлены либо участвующими структурами, включающими перегруппировку σ-электронов, либо специальным обозначением Y, имеющим три ядра в своих трех точках.
Делокализованные электроны важны по нескольким причинам; главная из них заключается в том, что ожидаемая химическая реакция может не произойти, потому что электроны делокализируются до более стабильной конфигурации, что приводит к реакции, которая происходит в другом месте. Примером может служить Friedel – Crafts алкилирование бензола с 1-хлор-2-метилпропаном; в карбокатион перестраивается в терт-бутил группа стабилизируется сверхсопряжение, особая форма делокализации. Делокализация приводит к удлинению длины волны электрона, следовательно, снижает энергию.
Бензол
Длина скрепления
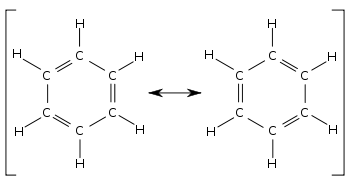
Сравнивая две составляющие структуры бензола, все одинарные и двойные связи меняются местами. Длина скрепления можно измерить, например, используя дифракция рентгеновских лучей. Средняя длина одинарной связи C – C составляет 154 вечера; двойная связь C = C составляет 133 пм. В локализованном циклогексатриене углерод-углеродные связи должны чередоваться 154 и 133 пм. Вместо этого все углерод-углеродные связи в бензоле имеют длину около 139 пм, что является промежуточным звеном между одинарной и двойной связью. Этот смешанный характер одинарной и двойной связи (или тройной связи) типичен для всех молекул, в которых связи имеют различный характер. ордер на облигации в различных способствующих структурах. Длину облигаций можно сравнить с помощью ордеров на облигации. Например, в циклогексане порядок связи равен 1, а в бензоле - 1 + (3 ÷ 6) =1 1⁄2. Следовательно, бензол имеет более характерную двойную связь и, следовательно, более короткую длину связи, чем циклогексан.
Резонансная энергия
Резонансная (или делокализационная) энергия - это количество энергии, необходимое для преобразования истинной делокализованной структуры в структуру наиболее стабильной вносящей вклад структуры. В эмпирическая резонансная энергия можно оценить путем сравнения изменение энтальпии из гидрирование реального вещества с расчетной структурой, вносящей вклад.
Полное гидрирование бензола до циклогексан через 1,3-циклогексадиен и циклогексен является экзотермический; 1 моль бензола дает 208,4 кДж (49,8 ккал).

Гидрирование одного моля двойных связей дает 119,7 кДж (28,6 ккал), как можно вывести из последней стадии, гидрирования циклогексена. В бензоле же 23,4 кДж (5,6 ккал) необходимо для гидрирования одного моля двойных связей. Разница, составляющая 143,1 кДж (34,2 ккал), представляет собой эмпирическую резонансную энергию бензола. Поскольку 1,3-циклогексадиен также имеет небольшую энергию делокализации (7,6 кДж или 1,8 ккал / моль), результирующая резонансная энергия по сравнению с локализованным циклогексатриеном немного выше: 151 кДж или 36 ккал / моль.[22]
Эта измеренная резонансная энергия также является разницей между энергией гидрирования трех «нерезонансных» двойных связей и измеренной энергией гидрирования:
- (3 × 119,7) - 208,4 = 150,7 кДж / моль (36 ккал).[23]
Квантово-механическое описание в теории VB

Резонанс имеет более глубокое значение в математическом формализме теория валентной связи (VB). Квантовая механика требует, чтобы волновая функция молекулы подчинялась наблюдаемой симметрии. Если единственная структура, способствующая этому, не достигает этого, вызывается резонанс.
Например, в бензоле теория валентных связей начинается с двух структур Кекуле, которые по отдельности не обладают шестикратной симметрией реальной молекулы. Теория конструирует действительные волновая функция как линейная суперпозиция волновых функций, представляющих две структуры. Поскольку обе структуры Кекуле имеют одинаковую энергию, они вносят равный вклад в общую структуру - суперпозиция представляет собой одинаково взвешенное среднее значение или линейную комбинацию 1: 1 двух в случае бензола. Симметричная комбинация дает основное состояние, а антисимметричная комбинация дает первое возбужденное состояние, как показано.
В общем случае суперпозиция записывается с неопределенными коэффициентами, которые затем вариативно оптимизированный найти минимально возможную энергию для заданного набора базисных волновых функций. Когда включается больше структур, вносящих вклад, молекулярная волновая функция становится более точной, и большее количество возбужденных состояний может быть получено из различных комбинаций структур, вносящих вклад.
Сравнение с теорией молекулярных орбиталей (МО)

В теория молекулярных орбиталей, основная альтернатива теория валентной связи, молекулярные орбитали (МО) аппроксимируются как суммы всех атомных орбиталей (АО) на всех атомах; Есть столько же МО, сколько и АО. Каждый АОя имеет взвешивание коэффициент cя что указывает на вклад АО в конкретную МО. Например, в бензоле модель МО дает нам 6 π МО, которые представляют собой комбинации 2pz АО на каждом из 6 атомов C. Таким образом, каждая π МО делокализована по всей молекуле бензола и любому электрону. занимающий МО будет делокализована по всей молекуле. Эта интерпретация МО послужила источником вдохновения для представления бензольного кольца в виде шестиугольника с кругом внутри. При описании бензола концепция локализованных σ-связей VB и концепция MO делокализованных π-орбиталей часто объединяются в курсах элементарной химии.
Содействующие структуры в модели VB особенно полезны при прогнозировании эффекта заместители на π-системах, таких как бензол. Они приводят к моделям способствующих структур для электроноакцепторная группа и электрон-выпускающая группа по бензолу. Полезность теории МО состоит в том, что количественный показатель заряда π-системы на атоме может быть получен из квадратов взвешивание коэффициент cя на атоме Cя. Обвинять qя ≈ c2
я. Причина возведения коэффициента в квадрат заключается в том, что если электрон описывается АО, то квадрат АО дает электронная плотность. AO настроены (нормализованный ) так что АО2 = 1 и qя ≈ (cяАОя)2 ≈ c2
я. В бензоле, qя = 1 на каждом атоме C. С электроноакцепторная группа qя <1 на орто и параграф Атомы углерода и qя > 1 для электрон-выпускающая группа.
Коэффициенты
Взвешивание участвующих структур с точки зрения их вклада в общую структуру можно рассчитать несколькими способами, используя "Ab initio" методы, основанные на теории Валентности Бонда или Орбитали естественной связи (NBO) подходы Weinhold NBO5 или, наконец, из эмпирических расчетов по методу Хюккеля. Программное обеспечение для обучения резонансу на основе метода Хюккеля доступно на HuLiS Интернет сайт.
Делокализация заряда
В случае ионов принято говорить о делокализованном заряде (делокализации заряда). Пример делокализованного заряда в ионах можно найти в карбоксилат группа, в которой отрицательный заряд одинаково центрирован на двух атомах кислорода. Делокализация заряда в анионах является важным фактором, определяющим их реакционную способность (обычно: чем выше степень делокализации, тем ниже реакционная способность) и, в частности, кислотная сила их сопряженных кислот. Как правило, чем лучше делокализован заряд в анионе, тем сильнее его конъюгированная кислота. Например, отрицательный заряд в перхлорат анион (ClO−
4) равномерно распределяется между симметрично ориентированными атомами кислорода (и часть его также удерживается центральным атомом хлора). Эта превосходная делокализация заряда в сочетании с большим количеством атомов кислорода (четыре) и высоким электроотрицательность центрального атома хлора приводит к хлорная кислота являясь одной из самых сильных известных кислот с pKа значение −10.[25]Степень делокализации заряда в анионе может быть количественно выражена с помощью параметра WAPS (средневзвешенная положительная сигма).[26] параметр и аналогичный WANS (средневзвешенная отрицательная сигма)[27][28] параметр используется для катионов.
| Сложный | WAPS × 105 | Сложный | WANS × 105 |
|---|---|---|---|
| (C2F5ТАК2)2NH | 2.0[29] | Трифенилфосфин | 2.1[27] |
| (CF3)3COH | 3.6[29] | Фенилтетраметилгуанидин | 2.5[27] |
| Пикриновая кислота | 4.3[26] | Трипропиламин | 2.6[27] |
| 2,4-динитрофенол | 4.9[26] | MTBD (7-метил-триазабициклодецен ) | 2.9[28] |
| Бензойная кислота | 7.1[26] | DBU (1,8-диазабициклоунд-7-ен ) | 3.0[28] |
| Фенол | 8.8[29] | TBD (Триазабициклодецен ) | 3.5[28] |
| Уксусная кислота | 16.1[26] | N,N-Диметиланилин | 4.7[27] |
| ЗДРАВСТВУЙ | 21.9[29] | Пиридин | 7.2[27] |
| HBr | 29.1[29] | Анилин | 8.2[27] |
| HCl | 35.9[26] | Пропиламин | 8.9[27] |
Значения WAPS и WANS приведены в е /Å4. Более высокие значения указывают на более локализованный заряд в соответствующем ионе.
Смотрите также
- Сопряженная система
- Делокализация
- Теория молекулярных орбиталей Хюккеля
- Гиперконъюгация
- Таутомерия
- Избегаемый переход
внешняя ссылка
- Goudard, N .; Carissan, Y .; Hagebaum-Reignier, D .; Хамбель, С. (2008). "HuLiS: Java-апплет - Простая теория Хюккеля и мезомерия - программное обеспечение для логики" (На французском). Получено 29 октября 2010.
Рекомендации
- ^ ИЮПАК, Сборник химической терминологии, 2-е изд. («Золотая книга») (1997). Исправленная онлайн-версия: (2006–) "Резонанс ". Дои:10.1351 / goldbook.R05326
- ^ ИЮПАК, Сборник химической терминологии, 2-е изд. («Золотая книга») (1997). Исправленная онлайн-версия: (2006–) "способствующая структура ". Дои:10.1351 / goldbook.C01309
- ^ а б Полинг, Линус (1960). «Понятие резонанса». Природа химической связи - введение в современную структурную химию (3-е изд.). Издательство Корнельского университета. С. 10–13. ISBN 978-0801403330.
- ^ Практикующие химики, знакомые с концепциями резонанса и делокализации, часто рисуют только одну основную структуру, которая неявно представляет молекулу, структура которой должна быть описана с помощью резонансного гибрида. Например, химик может произвольно нарисовать резонансную составляющую NO2– показано слева, при том понимании, что читатель знает другого участника, показанного справа, а также подразумевается, что связи N – O фактически эквивалентны. Эта практика особенно распространена в органической химии, где один из Кекуле структуры из бензол часто выбирают для изображения правильной гексагональной структуры молекулы.
- ^ Моррисон, Роберт; Бойд, Роберт (1989). «Глава 10». Органическая химия (5-е изд.). Прентис Холл Индии. п. 372. ISBN 978-0-87692-560-7.
Резонансный гибрид более стабилен, чем любая из составляющих его структур.
- ^ ИЮПАК, Сборник химической терминологии, 2-е изд. («Золотая книга») (1997).Исправленная онлайн-версия: (2006–) "резонансная энергия ". Дои:10.1351 / goldbook.R05333
- ^ «Резонансные формы». UCDavis Chem Вики. UCDavis. 2013-10-02. Получено 7 октября 2015.
- ^ Кербер, Роберт С. (2006). «Если это резонанс, то что резонирует?». J. Chem. Образовательный. 83 (2): 223. Bibcode:2006JChEd..83..223K. Дои:10.1021 / ed083p223.
- ^ «Графическое представление диаграмм химической структуры» (PDF), Рекомендации ИЮПАК 2008 г., ИЮПАК, п. 387 (GR – 8)
- ^ «Графическое представление диаграмм химической структуры» (PDF), Рекомендации ИЮПАК 2008 г., ИЮПАК, стр. 379–382 (GR – 6)
- ^ Тиле, Йоханнес (1899). "Zur Kenntnis der ungesättigten Verbindungen" [[Вклад] в наши знания о ненасыщенных соединениях]. Annalen der Chemie Юстуса Либиха (на немецком). 306: 87–142. Дои:10.1002 / jlac.18993060107. На стр. 89, Тиле ввел понятие «частичная валентность»: "Ich nehme nun an,… eine Partialvalens vorhanden ist, eine Annahme, die sich auch thermischrogründen lässt ». (Теперь я предполагаю, что в случае веществ, которым приписывается двойная связь, на самом деле для их связи используются два сродства каждого из участвующих атомов; однако, из-за способности к присоединению двойных связей, сила сродства не полностью расходуется, и в каждом из атомов существует остаток аффинности или «частичная валентность» - предположение, которое также может быть подтверждено термически (т.е. с помощью калориметрии). 90, Тиле ввел термин «сопряженный»: "Ein solches System benachbarter Doppelbindungen mit ausgeglichenen inneren Partialvalenzen sei als супружеская пара безеичнет ". (Такая система смежных двойных связей с уравновешенными внутренними частичными валентностями должна быть названа «сопряженной».) Тиле обсуждал сопряженную структуру бензола на стр. 125–129: VIII. Die aromatischen Verbindungen. Das Benzol. (VIII. Ароматические соединения. Бензол.)
- ^ Хорнбэк, Джозеф М. (2006). Органическая химия (2-е изд.). Томсон обучения. С. 470–1. ISBN 9780534389512.
- ^ Полинг, Линус, Резонанс, п. 1
- ^ "Наука и гуманизм Лайнуса Полинга". Архивировано 31 марта 2012 года.CS1 maint: BOT: статус исходного URL-адреса неизвестен (связь) См. Последний абзац раздела 1.
- ^ Полинг, Л. (1960). Природа химической связи (3-е изд.). Издательство Оксфордского университета. п.184. В этом источнике Полинг впервые упоминает связанные статьи Slater и Hückel в 1931 году, а затем цитирует свои собственные ключевые статьи: Полинг, Линус. (1931). "Природа химической связи. II. Одноэлектронная связь и трехэлектронная связь". Варенье. Chem. Soc. 53 (1367): 3225. Дои:10.1021 / ja01360a004. и последующие статьи 1932–1933 гг.
- ^ ИЮПАК, Сборник химической терминологии, 2-е изд. («Золотая книга») (1997). Исправленная онлайн-версия: (2006–) "Мезомерия ". Дои:10.1351 / goldbook.M03845
- ^ Мур, Баррингтон-младший (1954). Террор и прогресс СССР: некоторые источники перемен и стабильности в советской диктатуре. С. 142–143.
- ^ Литий всегда присутствует как Li+ (1 с2), дуэт, в ионных соединениях. В таких соединениях, как CH3Li с некоторой степенью ковалентности, связь достигается в основном с 2s-орбиталью, с некоторым вкладом от 2p-орбитали. (Эта схема связывания используется в агрегатах конденсированной фазы, таких как (CH3Ли)4 также, что приводит к более высокому координационному числу для лития.) Таким образом, в принципе может быть размещено до одного октета. Тем не менее, формальное количество валентных электронов вокруг Li никогда не превышает двух, если не включены слабые донорно-акцепторные взаимодействия с нейтральными лигандами (например, молекулы растворителя, часто опускаемые в структурах Льюиса).
- ^ Бречер, Джонатан (01.01.2008). «Стандарты графического представления диаграмм химической структуры (Рекомендации IUPAC 2008 г.)». Чистая и прикладная химия. 80 (2): 277–410. Дои:10.1351 / pac200880020277. ISSN 1365-3075.
- ^ Уэйд, Г. Органическая химия (6-е изд.).[ISBN отсутствует ]
- ^ Брюс, Паула Ю. Органическая химия (4-е изд.).[ISBN отсутствует ]
- ^ Виберг; Накаджи; Морган (1993). «Теплота гидрирования СНГ я добываю. Экспериментально-теоретическое исследование ». Варенье. Chem. Soc. 115 (9): 3527–3532. Дои:10.1021 / ja00062a017.
- ^ Шерман, Дж. (Февраль 1939 г.). «Теплоты гидрирования непредельных углеводородов». Варенье. Oil Chem. Soc. 16 (2): 28. Дои:10.1007 / BF02543208. S2CID 96029597. Архивировано из оригинал на 2011-07-14.
- ^ Shaik, Sason S .; Гиберти, Филипп С. (2008). Справочник химика по теории валентной связи. Нью-Джерси: Wiley-Interscience. стр.200 –203. ISBN 978-0-470-03735-5.
- ^ Продавцы, Кэтлин; Недели, Кэтрин; Олсоп, Уильям Р .; Клаф, Стивен Р .; Хойт, Мэрилин; Пью, Барбара (2006). Перхлорат: экологические проблемы и решения. CRC Press. п. 16. ISBN 978-0-8493-8081-5.
- ^ а б c d е ж Kaupmees, K .; Кальюранд, I .; Лейто, И. (2010). «Влияние содержания воды на кислотности в ацетонитриле. Количественная оценка делокализации заряда в анионах». J. Phys. Chem. А. 114 (43): 11788–11793. Bibcode:2010JPCA..11411788K. Дои:10.1021 / jp105670t. PMID 20919704.
- ^ а б c d е ж грамм час Kaupmees, K .; Кальюранд, I .; Лейто, И. (2014). «Влияние содержания воды на основность ацетонитрила». J. Solut. Chem. 43 (7): 1270–1281. Дои:10.1007 / s10953-014-0201-4. S2CID 95538780.
- ^ а б c d Kaupmees, K .; Trummal, A .; Лейто, И. (2014). «Основы сильных оснований в воде: вычислительное исследование». Хорват. Chem. Acta. 87 (4): 385–395. Дои:10.5562 / cca2472.
- ^ а б c d е Raamat, E .; Kaupmees, K .; Овсянников, Г .; Trummal, A .; Kütt, A .; Saame, J .; Koppel, I .; Кальюранд, I .; Губа, L .; Родима, Т .; Pihl, V .; Koppel, I.A .; Лейто, И. (2013). «Кислотности сильных нейтральных кислот Бренстеда в различных средах». J. Phys. Орг. Chem. 26 (2): 162–170. Дои:10.1002 / poc.2946.
| Авторитетный контроль |
|---|