Одноклеточный анализ - Single-cell analysis
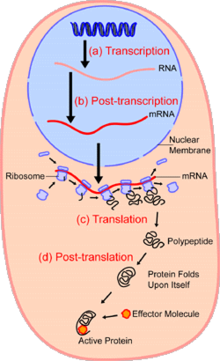
В областиклеточная биология, одноклеточный анализ это изучение геномика, транскриптомика, протеомика, метаболомика и межклеточные взаимодействия на уровне отдельной клетки.[1][2][3] Из-за гетерогенности, наблюдаемой как в популяциях эукариотических, так и в прокариотических клетках, анализ отдельной клетки позволяет обнаружить механизмы, которые не наблюдаются при изучении основной популяции клеток.[4] Такие технологии как сортировка клеток с активацией флуоресценции (FACS) позволяет точно изолировать отдельные отдельные ячейки от сложных образцов, в то время как высокопроизводительные технологии разделения отдельных ячеек,[5][6][7] возможность одновременного молекулярного анализа сотен или тысяч отдельных несортированных клеток; это особенно полезно для анализа вариации транскриптома в генотипически идентичных клетках, что позволяет определять подтипы клеток, которые иначе не обнаруживаются. Развитие новых технологий увеличивает нашу способность анализировать геном и транскриптом отдельных клеток, а также количественно определять их протеом и метаболом.[8][9][10] Методы масс-спектрометрии стали важными аналитическими инструментами для протеомного и метаболомного анализа отдельных клеток.[11][12] Недавние достижения позволили количественно оценить тысячи белков в сотнях отдельных клеток,[13] и, таким образом, делают возможными новые виды анализа.[14][15] Секвенирование in situ и флуоресценция in situ гибридизация (FISH) не требуют выделения клеток и все чаще используются для анализа тканей.[16]
Изоляция одной клетки
Многие методы анализа отдельных клеток требуют выделения отдельных клеток. Методы, используемые в настоящее время для выделения одиночных клеток, включают: диэлектрофорезную цифровую сортировку, ферментативное расщепление, FACS, гидродинамические ловушки, лазерная микродиссекция, ручной сбор, микрофлюидика, микроманипуляция, серийное разведение и рамановский пинцет.
Ручной сбор отдельных клеток - это метод, при котором клетки в суспензии просматриваются под микроскопом и выбираются индивидуально с помощью микропипетка.[17][18] Рамановский пинцет - это техника, в которойРамановская спектроскопия сочетается с оптический пинцет, который использует лазерный луч для улавливания и манипулирования клетками.[19]
В методе цифровой диэлектрофоретической сортировки используется массив электродов, управляемый полупроводником, в микрожидкостном чипе для улавливания отдельных клеток в диэлектрофорезных (DEP) клетках. Идентификация клеток обеспечивается комбинацией флуоресцентных маркеров с наблюдением изображения. Точная доставка обеспечивается управляемым полупроводником движением клеток DEP в проточной ячейке.
Разработка микрожидкостных биочипов на гидродинамической основе постоянно растет. В этом методе клетки или частицы захватываются в определенной области для анализа отдельных клеток (SCA), как правило, без какого-либо приложения внешних силовых полей, таких как оптические, электрические, магнитные или акустические. Существует необходимость изучить понимание SCA в естественном состоянии клетки, и разработка этих методов очень важна для этого исследования. Исследователи выделили огромное потенциальное поле, которое необходимо изучить для разработки биочипов, отвечающих требованиям рынка / исследователей. Гидродинамическая микрофлюидика облегчает разработку пассивных приложений «лаборатория на кристалле». В последнем обзоре содержится отчет о последних достижениях в этой области, а также об их механизмах, методах и приложениях.[20]
Связанные технологии
В методе цифровой диэлектрофоретической сортировки используется массив электродов, управляемый полупроводником, в микрожидкостном чипе для улавливания отдельных клеток в диэлектрофоретических (DEP) клетках. Идентификация клеток обеспечивается комбинацией флуоресцентных маркеров с наблюдением изображения. Точность доставки обеспечивается управляемым полупроводником движением клеток DEP в проточной ячейке.
Гидродинамические ловушки позволяют изолировать отдельную клетку в «ловушке» в одно и то же время с помощью пассивного микрофлюидного транспорта. Количество изолированных ячеек можно изменять в зависимости от количества ловушек в системе.
В методике микродиссекции с лазерным захватом используется лазер для рассечения и отделения отдельных клеток или срезов от образцов ткани, представляющих интерес. Эти методы включают наблюдение за клеткой под микроскопом, чтобы можно было идентифицировать и пометить участок для анализа, чтобы лазер мог разрезать клетку. Затем ячейку можно извлечь для анализа.
Ручной сбор отдельных клеток - это метод, при котором клетки в суспензии просматриваются под микроскопом и индивидуально собираются с помощью микропипетка.
Microfluidics позволяет изолировать отдельные клетки для дальнейшего анализа. Следующие принципы описывают различные микрофлюидные процессы для разделения отдельных клеток: изоляция на основе капель в масле, пневматические мембранные клапаны и гидродинамические ловушки для клеток. Микрожидкостные системы на основе капель в масле используют заполненные маслом каналы для удерживания разделенных капель воды. Это позволяет изолировать отдельную ячейку от каналов на масляной основе. Пневматические мембранные клапаны используют управление давлением воздуха для изоляции отдельных ячеек за счет отклонения мембраны. Манипулирование источником давления позволяет открывать или закрывать каналы в микрофлюидной сети. Обычно система требует оператора и имеет ограниченную пропускную способность.
Техника рамановского пинцета сочетает в себе использование Рамановская спектроскопия и оптический пинцет, которые используют лазерный луч для улавливания и манипулирования клетками.
Разработка микрожидкостных биочипов на гидродинамической основе постоянно растет. В этом методе клетки захватываются в определенной области для анализа отдельных клеток (SCA). Обычно это происходит без приложения внешних силовых полей, таких как оптическое, электрическое, магнитное или акустическое. Необходимо изучить особенности SCA в естественном состоянии клетки, и разработка этих методов очень важна для этого исследования. Исследователи подчеркнули необходимость разработки устройств на основе биочипов в соответствии с требованиями рынка и исследователей. Гидродинамическая микрофлюидика облегчает разработку пассивных приложений «лаборатория на кристалле».
Геномика
Методы
Одноклеточная геномика сильно зависит от увеличения количества копий ДНК, обнаруженных в клетке, поэтому их достаточно для секвенирования. Это привело к разработке стратегий для амплификация всего генома (WGA). В настоящее время стратегии WGA можно разделить на три категории:
- Контролируемое праймирование и ПЦР-амплификация: адаптер-линкер PCR WGA
- Случайное праймирование и ПЦР-амплификация: DOP-PCR, MALBAC
- Случайное праймирование и изотермическое усиление: MDA
Во многих сравнительных исследованиях сообщается, что адаптер Linker PCR WGA лучше всего подходит для анализа диплоидных мутаций единичных клеток благодаря очень низкому эффекту выпадения аллелей,[21][22][23] и для профилирования вариации числа копий из-за его низкого шума, как с aCGH, так и с NGS low Pass Sequencing.[24][25] Этот метод применим только к клеткам человека, как фиксированным, так и нефиксированным.
Один из широко используемых методов WGA называется полимеразной цепной реакцией с вырожденными олигонуклеотидами (DOP-PCR). В этом методе используется хорошо зарекомендовавший себя метод амплификации ДНК. ПЦР попытаться усилить весь геном, используя большой набор грунтовки. Несмотря на простоту, этот метод показал очень низкий охват генома. Усовершенствованием ДОФ-ПЦР является Многократное усиление смещения (MDA), в котором используются случайные праймеры и высокая точностьфермент, обычноДНК-полимераза Φ29, чтобы выполнить амплификацию более крупных фрагментов и больший охват генома, чем ДОФ-ПЦР. Несмотря на эти улучшения, MDA все еще имеет систематическую ошибку, зависящую от последовательности (некоторые части генома амплифицируются больше, чем другие из-за их последовательности). Показано, что метод, в значительной степени позволяющий избежать систематической ошибки, наблюдаемой при ДОФ-ПЦР и МДА, является Многократные циклы отжига и циклического усиления (МАЛЬБАК). Смещение в этой системе снижается только путем копирования исходной цепи ДНК вместо копирования копий. Основным недостатком использования MALBA является снижение точности по сравнению с DOP-PCR и MDA из-за фермента, используемого для копирования ДНК.[8] После амплификации с использованием любого из вышеперечисленных методов ДНК можно секвенировать с помощью Sanger или секвенирование следующего поколения (NGS).
Цель
Есть два основных приложения к изучению генома на уровне отдельной клетки. Одно из приложений - отслеживать изменения, происходящие в бактериальных популяциях, где часто наблюдаются фенотипические различия. Эти различия не учитываются при массовом секвенировании популяции, но могут наблюдаться при секвенировании отдельных клеток.[26] Второе важное приложение - изучение генетической эволюции рака. Поскольку раковые клетки постоянно мутируют, очень интересно посмотреть, как раковые клетки развиваются на генетическом уровне. Эти паттерны соматических мутаций и аберрации числа копий можно наблюдать с помощью секвенирования отдельных клеток.[1]
Транскриптомика
Методы
Транскриптомика одиночных клеток использует методы секвенирования, аналогичные геномике отдельных клеток, или прямое обнаружение с использованием флуоресценция in situ гибридизация. Первым шагом в количественной оценке транскриптома является преобразование РНК в кДНК с помощью обратная транскриптаза так что содержимое клетки можно секвенировать с использованием методов NGS, как это было сделано в геномике. После преобразования кДНК недостаточно для секвенирования, поэтому те же методы амплификации ДНК, которые обсуждаются в геномике отдельных клеток, применяются к кДНК, чтобы сделать секвенирование возможным.[1] В качестве альтернативы, флуоресцентные соединения, присоединенные к зондам гибридизации РНК, используются для идентификации конкретных последовательностей, а последовательное применение различных зондов РНК будет создавать всеобъемлющий транскриптом.[27][28]
Цель
Цель транскриптомики одиночных клеток - определить, какие гены экспрессируются в каждой клетке. Транскриптом часто используется для количественной оценки экспрессии гена вместо протеома из-за трудностей, связанных в настоящее время с увеличением уровней белка.[1]
Есть три основных причины, по которым экспрессия генов была изучена с использованием этого метода: изучение динамики генов, сплайсинг РНК и типирование клеток. Генную динамику обычно изучают, чтобы определить, какие изменения в экспрессии генов влияют на различные характеристики клеток. Например, этот тип транскриптомного анализа часто используется для изучения эмбрионального развития. Исследования сплайсинга РНК направлены на понимание регуляции различных транскрипционные изоформы. Транскриптомика отдельной клетки также использовалась для типирования клеток, когда гены, экспрессируемые в клетке, используются для идентификации типов клеток. Основная цель типизации ячеек - найти способ определить идентичность ячеек, о которых неизвестно. генетические маркеры.[1]
Протеомика
Методы
Существует три основных подхода к одноклеточной протеомике: методы на основе антител, методы на основе флуоресцентных белков и методы на основе масс-спектроскопии.[29][30]
Методы на основе антител
В методах, основанных на антителах, используются разработанные антитела для связывания с интересующими белками, что позволяет идентифицировать относительное количество множественных индивидуальных мишеней одним из нескольких различных методов.
Визуализация: Антитела могут быть связаны с флуоресцентными молекулами, такими как квантовые точки или помечены органическими флуорофоры для обнаружения флуоресцентная микроскопия. Поскольку к каждому антителу прикреплены квантовые точки разного цвета или уникальные флуорофоры, можно идентифицировать несколько разных белков в одной клетке. Квантовые точки можно смыть с антител, не повреждая образец, что позволяет проводить несколько циклов количественного определения белка с использованием этого метода на одном образце.[31] Для методов, основанных на органических флуорофорах, флуоресцентные метки прикрепляются с помощью обратимой связи, такой как ДНК-гибрид (который может быть расплавлен / диссоциирован в условиях с низким содержанием соли)[32] или химически инактивирован,[33] позволяет проводить несколько циклов анализа с количественным определением 3-5 целей за цикл. Эти подходы использовались для количественной оценки содержания белка в образцах биопсии пациента (например, рака) для картирования вариабельной экспрессии белка в тканях и / или опухолях,[33] и для измерения изменений в экспрессии белка и передаче клеточных сигналов в ответ на лечение рака.[32]
Массовая цитометрия: изотопы редких металлов, которые обычно не обнаруживаются в клетках или тканях, могут быть прикреплены к отдельным антителам и обнаружены с помощью масс-спектрометрии для одновременной и чувствительной идентификации белков.[34] Эти методы могут быть высоко мультиплексированы для одновременной количественной оценки многих мишеней (панели до 38 маркеров) в отдельных ячейках.[35]
Количественная оценка антитела-ДНК: другой метод, основанный на антителах, преобразует уровни белка в уровни ДНК.[29] Преобразование в ДНК позволяет увеличить уровни белка и использовать NGS для количественного определения белков. В одном из таких подходов выбирают два антитела для каждого белка, который необходимо определить количественно. Затем эти два антитела модифицируют, чтобы к ним была присоединена одноцепочечная ДНК, которая является комплементарной. Когда два антитела связываются с белком, комплементарные цепи отжигаются и образуют двухцепочечный сегмент ДНК, который затем можно амплифицировать с помощью ПЦР. Каждая пара антител, разработанная для одного белка, помечена другой последовательностью ДНК. Затем можно секвенировать ДНК, амплифицированную с помощью ПЦР, и количественно определить уровни белка.[36]
Методы на основе масс-спектроскопии
В протеомике, основанной на масс-спектроскопии, для идентификации пептидов необходимы три основных этапа: подготовка образца, разделение пептидов и идентификация пептидов. Несколько групп сосредоточили свое внимание на ооцитах или клетках на очень ранней стадии расщепления, поскольку эти клетки необычно велики и предоставляют достаточно материала для анализа.[37][38][39] Другой подход, протеомика отдельной клетки с помощью масс-спектрометрии (SCoPE-MS), позволил количественно определить тысячи белков в клетках млекопитающих с типичными размерами клеток (диаметр 10-15 мкм) путем комбинирования клеток-носителей и одноклеточного штрих-кодирования.[40][41][42][43] SCoPE-MS второго поколения,[44] SCoPE2,[45] увеличили производительность за счет автоматизированной и миниатюрной пробоподготовки;[43] одним из таких подходов является MAMS (Micro-Arrays for Mass Spectrometry), который обеспечивает аликвотирование на высоких скоростях за счет использования различий в смачиваемости между реципиентами и окружающими областями.[46] Он также улучшил количественную надежность и покрытие протеома за счет оптимизации ЖХ-МС / МС на основе данных.[47] и идентификация пептидов.[48] Существует несколько методов выделения пептидов для анализа. К ним относятся использование подготовка проб с помощью фильтров, использование магнитные бусины или с использованием ряда реагентов и этапов центрифугирования.[49][37][39] Разделение белков разного размера может быть выполнено с помощью капиллярный электрофорез (CE) или жидкостная хроматография (LC) (используя жидкостная хроматография с масс-спектроскопией также известен как ЖХ-МС).[37][38][39][40] Этот шаг упорядочивает пептиды перед количественным определением с использованием тандемная масс-спектроскопия (МС / МС). Основное различие между методами количественной оценки заключается в том, что некоторые используют метки на пептидах, такие как тандемные массовые теги (TMT) или диметиловые метки которые используются для определения того, из какой клетки произошел определенный белок (белки, поступающие из каждой клетки, имеют разные метки), в то время как другие не используют метки (количественно определяют клетки индивидуально). Затем данные масс-спектроскопии анализируются путем обработки данных через базы данных, которые преобразуют информацию об идентифицированных пептидах для количественного определения уровней белка.[37][38][39][40][50] Эти методы очень похожи на те, которые используются для количественно оценить протеом объемных клеток, с модификациями для очень небольшого объема образца.[41]
Цель
Цель изучения протеома - лучше понять активность клеток на уровне отдельных клеток. Поскольку белки отвечают за определение того, как действует клетка, понимание протеома отдельной клетки дает лучшее понимание того, как работает клетка и как изменяется экспрессия генов в клетке из-за различных стимулов окружающей среды. Хотя транскриптомика имеет ту же цель, что и протеомика, она не так точна при определении экспрессии генов в клетках, поскольку не принимает во внимание посттранскрипционная регуляция.[9] Транскриптомика по-прежнему важна, поскольку изучение разницы между уровнями РНК и белками может дать представление о том, какие гены регулируются посттранскрипционно.
Метаболомика
Методы
Существует четыре основных метода, используемых для количественной оценки метаболома отдельных клеток: детекция на основе флуоресценции, биосенсоры флуоресценции, FRET биосенсоры и масс-спектроскопия. Первые три перечисленных метода используют флуоресцентную микроскопию для обнаружения молекул в клетке. Обычно в этих анализах используются небольшие флуоресцентные метки, прикрепленные к интересующим молекулам, однако было показано, что это слишком инвазивно для метаболомики отдельных клеток и изменяет активность метаболитов. Текущее решение этой проблемы состоит в использовании флуоресцентных белков, которые будут действовать как детекторы метаболитов, флуоресцируя всякий раз, когда они связываются с интересующим метаболитом.[51]
Масс-спектроскопия становится наиболее часто используемым методом метаболомики отдельных клеток. Его преимущества заключаются в том, что нет необходимости разрабатывать флуоресцентные белки для всех представляющих интерес молекул, и он способен обнаруживать метаболиты в фемтомоль ассортимент.[12] Подобно методам, обсуждаемым в протеомике, также удалось добиться успеха в сочетании масс-спектроскопии с методами разделения, такими как капиллярный электрофорез, для количественного определения метаболитов. Этот метод также позволяет обнаруживать метаболиты в фемтомольных концентрациях.[51] Другой метод, использующий капиллярный микропробоотбор в сочетании с масс-спектрометрией с разделением ионной подвижности, показал, что он увеличивает молекулярный охват и разделение ионов для метаболомики отдельных клеток.[18][52] Исследователи пытаются разработать метод, который может удовлетворить то, чего не хватает нынешним методам: высокая пропускная способность, более высокая чувствительность для метаболитов, которые имеют меньшее количество или которые имеют низкую эффективность ионизации, хорошую воспроизводимость и позволяют количественно определять метаболиты.[53]
Цель
Цель метаболомики одиночных клеток состоит в том, чтобы лучше понять на молекулярном уровне основные биологические темы, такие как рак, стволовые клетки, старение, а также развитие лекарственной устойчивости. В целом метаболомика в основном сосредоточена на понимании того, как клетки справляются со стрессами окружающей среды на молекулярном уровне, и на предоставлении более динамичного понимания клеточных функций.[51]
Реконструкция траекторий развития
Транскриптомные анализы отдельных клеток позволили реконструировать траектории развития. Ветвление этих траекторий описывает дифференцировку клеток. Различные методы были разработаны для реконструкции ветвящихся траекторий развития на основе транскриптомных данных отдельных клеток.[54][55][56][57] Они используют различные передовые математические концепции из оптимальный транспорт[56] к главным графам.[57] Некоторые программные библиотеки для реконструкции и визуализации траекторий дифференциации клонов свободно доступны в Интернете.[58]
Межклеточное взаимодействие
Межклеточные взаимодействия характеризуются стабильными и временными взаимодействиями.
Смотрите также
использованная литература
- ^ а б c d е Ван Д., Бодовиц С. (июнь 2010 г.). "Анализ отдельных ячеек: новый рубеж в омике"'". Тенденции в биотехнологии. 28 (6): 281–90. Дои:10.1016 / j.tibtech.2010.03.002. ЧВК 2876223. PMID 20434785.
- ^ Хабиби И., Чеонг Р., Липняцки Т., Левченко А., Эмамян Е.С., Абди А. (апрель 2017 г.). «Вычисление и измерение ошибок принятия решений по ячейкам с использованием данных по одной ячейке». PLOS вычислительная биология. 13 (4): e1005436. Bibcode:2017PLSCB..13E5436H. Дои:10.1371 / journal.pcbi.1005436. ЧВК 5397092. PMID 28379950.
- ^ Merouane A, Rey-Villamizar N, Lu Y, Liadi I, Romain G, Lu J, et al. (Октябрь 2015 г.). «Автоматическое профилирование индивидуальных взаимодействий между клетками с помощью высокопроизводительной покадровой микроскопии изображений в решетках с нанолуками (TIMING)». Биоинформатика. 31 (19): 3189–97. Дои:10.1093 / биоинформатика / btv355. ЧВК 4693004. PMID 26059718.
- ^ Альтшулер С.Дж., Ву Л.Ф. (май 2010 г.). "Клеточная неоднородность: имеют ли различия значение?". Ячейка. 141 (4): 559–63. Дои:10.1016 / j.cell.2010.04.033. ЧВК 2918286. PMID 20478246.
- ^ Ху П, Чжан В, Синь Х, Дэн Г (2016-10-25). «Выделение и анализ отдельных клеток». Границы клеточной биологии и биологии развития. 4: 116. Дои:10.3389 / fcell.2016.00116. ЧВК 5078503. PMID 27826548.
- ^ Мора-Кастилья С., То С, Ваезеслами С., Мори Р., Сринивасан С., Дамди Дж. Н. и др. (Август 2016 г.). «Технологии миниатюризации для эффективной подготовки одноклеточных библиотек для секвенирования следующего поколения». Журнал автоматизации лабораторий. 21 (4): 557–67. Дои:10.1177/2211068216630741. ЧВК 4948133. PMID 26891732.
- ^ Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, et al. (Январь 2017 г.). «Массивно-параллельное цифровое транскрипционное профилирование отдельных клеток». Nature Communications. 8: 14049. Bibcode:2017НатКо ... 814049Z. Дои:10.1038 / ncomms14049. ЧВК 5241818. PMID 28091601.
- ^ а б Хуан Л., Ма Ф, Чепмен А., Лу С., Се XS (2015). «Одноклеточная амплификация всего генома и секвенирование: методология и приложения». Ежегодный обзор геномики и генетики человека. 16 (1): 79–102. Дои:10.1146 / annurev-genom-090413-025352. PMID 26077818. S2CID 12987987.
- ^ а б Ву А.Р., Ван Дж., Улицы А.М., Хуан Ю. (июнь 2017 г.). «Транскрипционный анализ одиночных клеток». Ежегодный обзор аналитической химии. 10 (1): 439–462. Дои:10.1146 / annurev-anchem-061516-045228. PMID 28301747. S2CID 40069109.
- ^ Циорис К., Торрес А.Дж., Дус ТБ, Лав Дж.С. (2014). «Новый набор инструментов для оценки отдельных ячеек». Ежегодный обзор химической и биомолекулярной инженерии. 5: 455–77. Дои:10.1146 / annurev-chembioeng-060713-035958. ЧВК 4309009. PMID 24910919.
- ^ Comi TJ, Do TD, Rubakhin SS, Sweedler JV (март 2017). «Категоризация клеток на основе их химических профилей: прогресс в масс-спектрометрии одиночных клеток». Журнал Американского химического общества. 139 (11): 3920–3929. Дои:10.1021 / jacs.6b12822. ЧВК 5364434. PMID 28135079.
- ^ а б Чжан Л., Вертес А. (апрель 2018 г.). «Подходы одноклеточной масс-спектрометрии к исследованию клеточной гетерогенности». Angewandte Chemie. 57 (17): 4466–4477. Дои:10.1002 / anie.201709719. PMID 29218763. S2CID 4928231.
- ^ Славов Н (июнь 2020). «Одноклеточный анализ белков с помощью масс-спектрометрии». Современное мнение в области химической биологии. 60: 1–9. arXiv:2004.02069. Дои:10.1016 / j.cbpa.2020.04.018. PMID 32599342. S2CID 219966629.
- ^ Шпехт Х, Славов Н (август 2018). «Трансформирующие возможности одноклеточной протеомики». Журнал протеомных исследований. 17 (8): 2565–2571. Дои:10.1021 / acs.jproteome.8b00257. ЧВК 6089608. PMID 29945450.
- ^ Славов Н (январь 2020 г.). «Открытие протеома в отдельных клетках». Наука. 367 (6477): 512–513. Bibcode:2020Sci ... 367..512S. Дои:10.1126 / science.aaz6695. ЧВК 7029782. PMID 32001644.
- ^ Ли Дж. Х. (июль 2017 г.). «Реконструкция экспрессии гена De Novo в космосе». Тенденции в молекулярной медицине. 23 (7): 583–593. Дои:10.1016 / j.molmed.2017.05.004. ЧВК 5514424. PMID 28571832.
- ^ Гросс А., Шендубе Дж., Циммерманн С., Стиб М., Зенгерле Р., Колтай П. (июль 2015 г.). «Технологии одиночной изоляции». Международный журнал молекулярных наук. 16 (8): 16897–919. Дои:10.3390 / ijms160816897. ЧВК 4581176. PMID 26213926.
- ^ а б Чжан Л., Вертес А. (октябрь 2015 г.). «Энергетический заряд, окислительно-восстановительное состояние и обмен метаболитов в единичных гепатоцитах человека, выявленные с помощью масс-спектрометрии с капиллярным микровыбором». Аналитическая химия. 87 (20): 10397–405. Дои:10.1021 / acs.analchem.5b02502. PMID 26398405.
- ^ Фариа Э, Гарднер П (01.01.2012). Линдстрём С., Андерссон-Сван Х (ред.). Одноклеточный анализ. Методы молекулярной биологии. 853. Humana Press. С. 151–167. Дои:10.1007/978-1-61779-567-1_12. ISBN 9781617795664. PMID 22323146.
- ^ Нараянамурти В., Нагараджан С., Хан А.Ю., Самсури Ф., Шридхар Т.М. (30.06.2017). «Микрожидкостный гидродинамический захват для анализа отдельных клеток: механизмы, методы и приложения». Аналитические методы. 9 (25): 3751–3772. Дои:10.1039 / C7AY00656J. ISSN 1759-9679.
- ^ Бабаян А., Алави М., Гормли М., Мюллер В., Викман Х., МакМаллин Р.П. и др. (Август 2017 г.). «Сравнительное исследование характеристик амплификации всего генома и секвенирования нового поколения единичных раковых клеток». Oncotarget. 8 (34): 56066–56080. Дои:10.18632 / oncotarget.10701. ЧВК 5593545. PMID 28915574.
- ^ Биндер В., Бартенхаген С., Окпаньи В., Гомберт М., Мёлендик Б., Беренс Б. и др. (Октябрь 2014 г.). «Новый рабочий процесс для полногеномного секвенирования отдельных клеток человека». Человеческая мутация. 35 (10): 1260–70. Дои:10.1002 / humu.22625. PMID 25066732. S2CID 27392899.
- ^ Боргстрём Э, Патерлини М, Молд Дж. Э., Фризен Дж., Лундеберг Дж. (2017). «Сравнение методов амплификации всего генома для секвенирования экзома одной клетки человека». PLOS ONE. 12 (2): e0171566. Bibcode:2017PLoSO..1271566B. Дои:10.1371 / journal.pone.0171566. ЧВК 5313163. PMID 28207771.
- ^ Normand E, Qdaisat S, Bi W, Shaw C, Van den Veyver I, Beaudet A, Breman A (сентябрь 2016 г.). «Сравнение трех методов полногеномной амплификации для обнаружения геномных аберраций в отдельных клетках». Пренатальная диагностика. 36 (9): 823–30. Дои:10.1002 / pd.4866. PMID 27368744. S2CID 5537482.
- ^ Vander Plaetsen AS, Deleye L, Cornelis S, Tilleman L, Van Nieuwerburgh F, Deforce D (декабрь 2017 г.). «STR-профилирование и анализ вариации числа копий на отдельных сохраненных клетках с использованием современных методов амплификации всего генома». Научные отчеты. 7 (1): 17189. Bibcode:2017НатСР ... 717189В. Дои:10.1038 / s41598-017-17525-5. ЧВК 5719346. PMID 29215049.
- ^ Калиский Т., Quake SR (апрель 2011 г.). «Одноклеточная геномика». Методы природы. 8 (4): 311–4. Дои:10.1038 / nmeth0411-311. PMID 21451520. S2CID 5601612.
- ^ Любек Э., Джошкун А.Ф., Жиентаев Т., Ахмад М., Цай Л. (апрель 2014 г.). «Профилирование РНК in situ одной клетки путем последовательной гибридизации». Методы природы. 11 (4): 360–1. Дои:10.1038 / nmeth.2892. ЧВК 4085791. PMID 24681720.
- ^ Чен К.Х., Боеттигер А.Н., Моффитт-младший, Ван С., Чжуан Х (апрель 2015 г.). «Визуализация РНК. Пространственно разрешенное, высоко мультиплексное профилирование РНК в отдельных клетках» (PDF). Наука. 348 (6233): ааа6090. Дои:10.1126 / science.aaa6090. ЧВК 4662681. PMID 25858977.
- ^ а б Леви Э., Славов Н (октябрь 2018 г.). «Анализ белков отдельных клеток для системной биологии». Очерки биохимии. 62 (4): 595–605. Дои:10.1042 / EBC20180014. ЧВК 6204083. PMID 30072488.
- ^ Славов Н (январь 2020 г.). «Открытие протеома в отдельных клетках». Наука. 367 (6477): 512–513. Bibcode:2020Sci ... 367..512S. Дои:10.1126 / science.aaz6695. ЧВК 7029782. PMID 32001644.
- ^ Зражевский П., True LD, Gao X (октябрь 2013 г.). «Многоцветное многоцикловое молекулярное профилирование с квантовыми точками для анализа отдельных клеток». Протоколы природы. 8 (10): 1852–69. Дои:10.1038 / nprot.2013.112. ЧВК 4108347. PMID 24008381.
- ^ а б Giedt RJ, Pathania D, Carlson JC, McFarland PJ, Del Castillo AF, Juric D, Weissleder R (октябрь 2018 г.). «Анализ одноклеточного штрих-кода обеспечивает быстрое считывание клеточных сигнальных путей в клинических образцах». Nature Communications. 9 (1): 4550. Bibcode:2018НатКо ... 9,4550 г. Дои:10.1038 / s41467-018-07002-6. ЧВК 6208406. PMID 30382095.
- ^ а б Лин Дж. Р., Изар Б., Ван С., Япп С., Мей С., Шах П. М. и др. (Июль 2018). Чакраборти А.К., Радж А., Марр С., Хорват П. (ред.). «Высоко мультиплексная иммунофлуоресцентная визуализация тканей и опухолей человека с использованием t-CyCIF и обычных оптических микроскопов». eLife. 7: e31657. Дои:10.7554 / eLife.31657. ЧВК 6075866. PMID 29993362.
- ^ Наир Н., Мей Х.Э., Чен С.Ю., Хейл М., Нолан Г.П., Маеккер Х.Т. и др. (Май 2015 г.). «Массовая цитометрия как платформа для открытия клеточных биомаркеров для эффективного лечения ревматических заболеваний». Исследования и лечение артрита. 17: 127. Дои:10.1186 / s13075-015-0644-z. ЧВК 4436107. PMID 25981462.
- ^ Спитцер М.Х., Нолан Г.П. (май 2016 г.). «Массовая цитометрия: отдельные клетки, многие особенности». Ячейка. 165 (4): 780–91. Дои:10.1016 / j.cell.2016.04.019. ЧВК 4860251. PMID 27153492.
- ^ Гонг Х., Холкомб И., Оои А., Ван Х, Маджонис Д., Унгер М.А., Рамакришнан Р. (январь 2016 г.). «Простой метод получения антител, конъюгированных с олигонуклеотидами, и его применение при обнаружении мультиплексных белков в одиночных клетках». Биоконъюгат Химия. 27 (1): 217–25. Дои:10.1021 / acs.bioconjchem.5b00613. PMID 26689321.
- ^ а б c d Ломбард-Банек С., Редди С., Муди С.А., Немес П. (август 2016 г.). «Безмаркировочная количественная оценка белков в одиночных эмбриональных клетках с нервной судьбой в эмбрионе лягушки на стадии дробления (Xenopus laevis) с использованием капиллярного электрофореза с ионизацией электрораспылением с высокой разрешающей способностью масс-спектрометрии (CE-ESI-HRMS)». Молекулярная и клеточная протеомика. 15 (8): 2756–68. Дои:10.1074 / mcp.M115.057760. ЧВК 4974349. PMID 27317400.
- ^ а б c Сунь Л., Дубяк К.М., Пеучен Э.Х., Чжан З., Чжу Г., Хубер П.В., Довичи, штат Нью-Джерси (июль 2016 г.). «Протеомика одиночных клеток с использованием бластомеров лягушки (Xenopus laevis), выделенных из зародышей на ранних стадиях, которые формируют геометрическую прогрессию содержания белка». Аналитическая химия. 88 (13): 6653–7. Дои:10.1021 / acs.analchem.6b01921. ЧВК 4940028. PMID 27314579.
- ^ а б c d Вирант-Клун И., Лейхт С., Хьюз С., Крайгсвельд Дж. (Август 2016 г.). «Идентификация белков, специфичных для созревания, с помощью одноклеточной протеомики ооцитов человека». Молекулярная и клеточная протеомика. 15 (8): 2616–27. Дои:10.1074 / mcp.M115.056887. ЧВК 4974340. PMID 27215607.
- ^ а б c Будник Б, Леви Э, Славов Н (2017-03-15). «Масс-спектрометрия отдельных клеток млекопитающих количественно определяет гетерогенность протеома во время дифференцировки клеток». bioRxiv 10.1101/102681.
- ^ а б "SCoPE-MS - Наконец-то мы можем заняться протеомикой одиночных клеток !!!". Новости протеомических исследований. 2017-03-09. Получено 2017-06-28.
- ^ «Одноклеточная протеомика - блог Slavov Lab». Блог Slavov Lab. 2017-06-06. Получено 2017-06-27.
- ^ а б Specht H, Harmange G, Perlman DH, Emmott E, Niziolek Z, Budnik B, Slavov N (2018-08-25). «Автоматизированная пробоподготовка для высокопроизводительной одноклеточной протеомики». bioRxiv: 399774. Дои:10.1101/399774.
- ^ Будник Б., Леви Е., Харманге Г., Славов Н. (октябрь 2018 г.). «SCoPE-MS: масс-спектрометрия отдельных клеток млекопитающих количественно определяет гетерогенность протеома во время дифференцировки клеток». Геномная биология. 19 (1): 161. Дои:10.1186 / s13059-018-1547-5. ЧВК 6196420. PMID 30343672.
- ^ Шпехт Х, Эммотт Э, Коллер Т, Славов Н (2019-07-09). «Высокопроизводительная одноклеточная протеомика количественно определяет возникновение неоднородности макрофагов». bioRxiv. Дои:10.1101/665307.
- ^ Urban PL, Jefimovs K, Amantonico A, Fagerer SR, Schmid T, Mädler S и др. (Декабрь 2010 г.). «Микромассивы высокой плотности для масс-спектрометрии». Лаборатория на чипе. 10 (23): 3206–9. Дои:10.1039 / C0LC00211A. PMID 20938499. S2CID 8747868.
- ^ Хаффман Р.Г., Чен А., Шпехт Х., Славов Н. (июнь 2019). "DO-MS: Оптимизация методов масс-спектрометрии на основе данных". Журнал протеомных исследований. 18 (6): 2493–2500. Дои:10.1021 / acs.jproteome.9b00039. ЧВК 6737531. PMID 31081635.
- ^ Чен А.Т., Франкс А., Славов Н. (июль 2019 г.). Кокс Дж (ред.). «DART-ID увеличивает покрытие протеома одной клетки». PLOS вычислительная биология. 15 (7): e1007082. Bibcode:2019PLSCB..15E7082C. Дои:10.1371 / journal.pcbi.1007082. ЧВК 6625733. PMID 31260443.
- ^ Вишневски Дж. Р., Зугман А., Нагарадж Н., Манн М. (май 2009 г.). «Универсальный метод пробоподготовки для протеомного анализа». Методы природы. 6 (5): 359–62. Дои:10.1038 / nmeth.1322. PMID 19377485. S2CID 205418951.
- ^ Смитс AH, Lindeboom RG, Perino M, van Heeringen SJ, Veenstra GJ, Vermeulen M (сентябрь 2014 г.). «Глобальная абсолютная количественная оценка показывает жесткую регуляцию экспрессии белка в отдельных яйцах Xenopus». Исследования нуклеиновых кислот. 42 (15): 9880–91. Дои:10.1093 / нар / gku661. ЧВК 4150773. PMID 25056316.
- ^ а б c Зеноби Р. (декабрь 2013 г.). «Метаболомика одноклеточной: аналитические и биологические перспективы». Наука. 342 (6163): 1243259. Дои:10.1126 / science.1243259. PMID 24311695. S2CID 21381091.
- ^ Чжан Л., Форман Д.П., Грант П.А., Шреста Б., Муди С.А., Вильерс Ф. и др. (Октябрь 2014 г.). «Метаболический анализ in situ отдельных растительных клеток с помощью капиллярного микровыбора проб и масс-спектрометрии с ионизацией электрораспылением с разделением подвижности ионов». Аналитик. 139 (20): 5079–85. Bibcode:2014Ana ... 139.5079Z. Дои:10.1039 / C4AN01018C. PMID 25109271.
- ^ Дункан К.Д., Фирестам Дж., Ланекофф И. (январь 2019 г.). «Достижения в метаболомике единичных клеток на основе масс-спектрометрии». Аналитик. 144 (3): 782–793. Bibcode:2019Ana ... 144..782D. Дои:10.1039 / C8AN01581C. PMID 30426983.
- ^ Haghverdi L, Büttner M, Wolf FA, Buettner F, Theis FJ (октябрь 2016 г.). «Диффузионное псевдотовремя надежно реконструирует ветвление клонов» (PDF). Методы природы. 13 (10): 845–8. Дои:10.1038 / nmeth.3971. PMID 27571553. S2CID 3594049.
- ^ Сетти М. и др. Wishbone идентифицирует бифуркационные траектории развития на основе данных одной клетки. Nat. Biotechnol. 34. С. 637–645 (2016).
- ^ а б Schiebinger G, Shu J, Tabaka M, Cleary B, Subramanian V, Solomon A, et al. (Февраль 2019). «Оптимальный транспортный анализ экспрессии гена одной клетки определяет траектории развития при репрограммировании». Ячейка. 176 (4): 928–943.e22. Дои:10.1016 / j.cell.2019.01.006. ЧВК 6402800. PMID 30712874.
- ^ а б Chen H, Albergante L, Hsu JY, Lareau CA, Lo Bosco G, Guan J и др. (Апрель 2019 г.). «Реконструкция траекторий отдельных ячеек, исследование и отображение данных omics с помощью STREAM». Nature Communications. 10 (1): 1903. Bibcode:2019НатКо..10.1903C. Дои:10.1038 / s41467-019-09670-4. ЧВК 6478907. PMID 31015418.
- ^ Pinello Lab. Исследование и картографирование реконструкции траектории одной ячейки