Химическое гликозилирование - Chemical glycosylation
А реакция химического гликозилирования предполагает соединение гликозильный донор, в гликозильный акцептор формирование гликозид.[1][2][3] Если и донор, и акцептор являются сахарами, то продукт является олигосахарид. Реакция требует активации подходящим активирующим реагентом. Реакции часто приводят к смешиванию продуктов из-за создания нового стереогенный центр на аномерное положение донора гликозила. Формирование гликозидная связь позволяет синтезировать сложные полисахариды которые могут играть важную роль в биологических процессах и патогенез и поэтому наличие синтетических аналогов этих молекул позволяет проводить дальнейшие исследования в отношении их биологического значения.
Терминология
Реакция гликозилирования включает связывание гликозильного донора и гликозильного акцептора посредством инициирования с использованием активатора в подходящих условиях реакции.
- А гликозильный донор представляет собой сахар с подходящей уходящей группой в аномерном положении. Эта группа в условиях реакции активируется и через образование оксокарбений устраняется, оставляя электрофильный аномерный углерод.
- А гликозильный акцептор сахар с незащищенным нуклеофильный гидроксильная группа которые могут атаковать углерод иона оксокарбения, образующегося в ходе реакции, и способствовать образованию гликозидной связи.
Активатором обычно является Кислота Льюиса что позволяет уходящей группе в аномерном положении уходить и приводит к образованию иона оксокарбения.
Стереохимия
Образование гликозидной связи приводит к образованию нового стереогенный центр и поэтому можно ожидать, что в результате получится смесь продуктов. Образованная связь может быть аксиальной или экваториальной (α или β по отношению к глюкозе). Чтобы лучше понять это, необходимо рассмотреть механизм реакции гликозилирования.

Участие в соседней группе
Стереохимический результат реакции гликозилирования может в некоторых случаях зависеть от типа защитной группы, используемой в положении 2 гликозильного донора. Участвующая группа, обычно одна с присутствующей карбоксильной группой, будет преимущественно приводить к образованию β-гликозида. В то время как неучаствующая группа, группа, обычно не имеющая карбоксильной группы, часто приводит к α-гликозиду.
Ниже можно увидеть, что наличие ацетильной защитной группы в положении 2 позволяет образовывать ион ацетоксония промежуточное соединение, которое блокирует атаку на нижнюю поверхность кольца, что позволяет преимущественно образовывать β-гликозид.
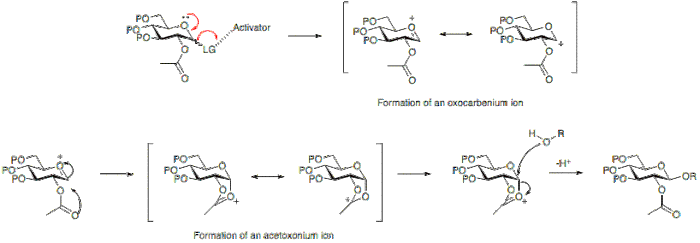
В качестве альтернативы, отсутствие участвующей группы в позиции 2 позволяет атаковать либо снизу, либо сверху. Поскольку α-гликозидный продукт будет предпочтительнее аномерный эффект, обычно преобладает α-гликозид.

Защита групп
Другой защитные группы на гликозильном доноре или гликозильном акцепторе[4][5] может влиять на реакционную способность и выход реакции гликозилирования. Обычно электроноакцепторные группы такие как ацетильная или бензоильная группы, как обнаружено, уменьшают реакционную способность донора / акцептора и поэтому называются «обезоруживающими» группами. Электронно-донорские группы такие как бензильная группа, как обнаружено, увеличивают реактивность донора / акцептора и поэтому называются «активирующими» группами.
Современные методы синтеза гликозидов
Гликозилиодиды
Гликозилиодиды были впервые введены для использования в реакциях гликозилирования в 1901 г. Кенигс и Кнорр[6][7] хотя часто считались слишком реактивными для синтетического использования. Недавно несколько исследовательских групп показали, что эти доноры обладают уникальными реакционными свойствами и могут отличаться от других гликозилхлоридов или бромидов в отношении времени реакции, эффективности и стереохимия.[8][9][10][11] Гликозилиодиды могут быть получены в различных условиях, одним из примечательных методов является реакция 1-О-ацетилпиранозид с TMSI.[12]

Доноры йодида обычно можно активировать в основных условиях с получением β-гликозидов с хорошей селективностью. Использование йодидных солей тетраалкиламмония, таких как йодид тетрабутиламмония (TBAI ) позволяет на месте аномеризация от α-гликозилгалогенида к β-гликозилгалогениду и обеспечивает α-гликозид с хорошей селективностью.[13][14][15][16]

Тиогликозиды
Тиогликозиды впервые были описаны в 1909 г. Фишер [17] и с тех пор постоянно исследовались, что позволило разработать многочисленные протоколы их получения. Преимущество использования тиогликозидов заключается в их стабильности в широком диапазоне условий реакции, позволяющих манипулировать защитными группами. Кроме того, тиогликозиды действуют как временные защитные группы в аномерном положении, позволяя тиогликозидам быть полезными как в качестве доноров гликозила, так и в качестве акцепторов гликозила.[13]Тиогликозиды обычно получают реакцией перацетилированных сахаров с BF.3• OEt2 и соответствующий тиол.[18][19][20]

Тиогликозиды, используемые в реакциях гликозилирования в качестве доноров, могут быть активированы в широком диапазоне условий, в первую очередь с использованием NIS / AgOTf.[21]

Трихлорацетимидаты
Трихлорацетимидаты были впервые представлены и исследованы Шмидтом в 1980 г.[22][23] и с тех пор стали очень популярными для синтеза гликозидов. Использование трихлорацетимидатов дает множество преимуществ, включая простоту образования, реакционную способность и стереохимический результат.[13] О-Гликозилтрихлорацетимидаты получают путем добавления трихлорацетонитрил (Cl3CCN) в основных условиях к свободной аномерной гидроксильной группе.

Типичными активирующими группами для реакций гликозилирования с использованием трихлорацетимидатов являются BF3• OEt2 или TMSOTf.[24]

Колоночная хроматографическая очистка реакционной смеси иногда может быть сложной задачей из-за побочного продукта - трихлорацетамида. Однако этого можно избежать, промывая органический слой 1 М раствором NaOH в делительной воронке перед хроматографией. Было обнаружено, что защитные группы ацетила стабильны во время этой процедуры.[25]
Известные синтетические продукты
Ниже приведены несколько примеров некоторых известных мишеней, полученных с помощью серии реакций гликозилирования.


Смотрите также
- Гликозилирование
- Гликозилтрансфераза
- Гликорандомизация
- Гликирование
- Углеводная химия
- Олигосахарид
- Углеводы
использованная литература
- ^ Бунс, Герт-Ян; Карл Дж. Хейл (2000). Органический синтез с углеводами. Блэквелл Паблишинг. ISBN 978-1-85075-913-3.
- ^ Crich, D .; Лим, Л. Орг. Реагировать. 2004, 64, 115. Дои:10.1002 / 0471264180.or064.02
- ^ Буфали, С .; Зеебергер, П. Орг. Реагировать. 2006, 68, 303. Дои:10.1002 / 0471264180.or064.02
- ^ Ворм, Стефан ван дер; Хансен, Томас; Хенгст, Якоб М.А. ван; С. Оверклефт, Герман; Marel, Gijsbert A. van der; К. Коде, Джерун Д. (2019). «Реактивность акцептора в реакциях гликозилирования». Обзоры химического общества. 48 (17): 4688–4706. Дои:10.1039 / C8CS00369F. PMID 31287452.
- ^ Vorm, S. van der; Hansen, T .; S. Overkleeft, H .; Marel, G.A. van der; К. Коде, Дж. Д. (2017). «Влияние нуклеофильности акцептора на механизм реакции гликозилирования». Химическая наука. 8 (3): 1867–1875. Дои:10.1039 / C6SC04638J. ЧВК 5424809. PMID 28553477.
- ^ Вильгельм Кенигс и Эдвард Норр (1901). "Ueber einige Derivate des Traubenzuckers und der Galactose (p)". Berichte der deutschen chemischen Gesellschaft. 34 (1): 957–981. Дои:10.1002 / cber.190103401162.
- ^ Фишер, Э. Бер. Dtsch. Chem. Ges. 1893, 26, 2400–2412
- ^ Гервей Дж., Хадд М. Дж., J. Org. Chem. 1997, 62, 6961 – 6967.
- ^ Хадд, М. Дж., Джервей, Дж. Carbohydr. Res. 1999, 320, 61 – 69.
- ^ Микель, Н., Виньяндо, С., Руссо, Г., Лэй, Л. Synlett 2004, 2, 341 – 343.
- ^ ван Велл, Р. М., Карта, К. П. Р., Филд, Р. А. J. Carbohydr. Chem. 2005, 24, 463 – 474.
- ^ Джервей, Дж., Нгуен, Т. Н., Хадд, М. Дж. Carbohydr. Res. 1997, 300, 119 – 125.
- ^ а б c Чжу, Х.М., Шмидт, Р.Р. Энгью. Chem. Int. Эд. 2009, 48, 1900 - 1934.
- ^ Лам, С. Н., Жервей-Гааг, Дж. Орг. Lett. 2003, 5, 4219 – 4222.
- ^ Du, W., Gervay-Hague, J. Орг. Lett. 2005, 7, 2063–2065.
- ^ Du, W., Kulkarni, S. S., Gervay-Hague, J. Chem. Commun. 2007, 2336–2338.
- ^ Фишер, Э., Дельбрюк, К. Бер. Dtsch. Chem. Ges. 1909, 42, 1476–1482.
- ^ Тай, К.А., Кулкарни, С.С., Хунг, С.С. J. Org.Chem. 2003, 68, 8719 – 8722
- ^ Агнихотри, Г., Тивари, П., Мисра, А. К. Carbohydr. Res. 2005, 340, 1393–1396
- ^ Хасэ-гава, Дж. Й., Хамада, М., Миямото, Т., Нисиде, К., Каджимото, Т., Уениши, Дж. И., Узел, М. Carbohydr. Res. 2005, 340, 2360–2368.
- ^ Винеман, Г. Х., ван Леувен, С. Х., ван Бум, Дж. Х. Tetrahedron Lett. 1990, 31, 1331–1334.
- ^ Шмидт, Р. Р., Мишель, Дж. Энгью. Chem. 1980, 92, 763 – 764.
- ^ Шмидт, Р. Р., Мишель, Дж. Энгью. Chem. Int. Эд. Англ. 1980, 19, 731 – 732.
- ^ Кейл, Р. Р., МакГаннон, К. М., Фуллер-Шефер, К., Хэтч, Д. М., Флаглер, М. Дж., Гэмидж, С. Д., Вайс, А. А., Айер, С. С. Энгью. Chem. Int. Эд. 2008, 47, 1265 - 1268.
- ^ Хейкендорф, Мэдс; Дженсен, Хенрик Х. (01.02.2017). «Удаление некоторых распространенных побочных продуктов гликозилирования во время обработки реакции». Исследование углеводов. 439: 50–56. Дои:10.1016 / j.carres.2016.12.007. ISSN 0008-6215.
- ^ Джо, М., Бай, Ю., Накарио, Р. К., Ловари, Т. Л. Варенье. Chem. Soc. 2007, 129, 9885 - 9901.
- ^ Ву, X. Y., Бандл, Д. Р. J. Org. Chem. 2005, 70, 7381 - 7388.
- Марко Брито-Ариас, Синтез и характеристика гликозидов, второе издание, издание Springer, 2016 г.