Энамин - Enamine - Wikipedia

An енамин является ненасыщенное соединение полученный конденсацией альдегид или же кетон со вторичным амин.[1][2] Енамины - это универсальные промежуточные продукты.[3][4]
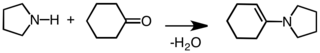 Конденсация с образованием енамина.[5]
Конденсация с образованием енамина.[5]
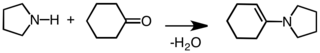
Слово «енамин» образовано от аффикса en-, используется как суффикс алкен, а корень амин. Это можно сравнить с энол, которая представляет собой функциональную группу, содержащую как алкен (en-) и алкоголь (-ол). Енамины считаются азотными аналогами енолов.[6]
Если один из заместителей азота представляет собой атом водорода, H, это таутомерный форма я добываю. Обычно это перестраивается в имин; однако есть несколько исключений (например, анилин ). Енамин-иминную таутомерию можно считать аналогом кето-енольная таутомерия. В обоих случаях атом водорода меняет свое положение между гетероатомом (кислородом или азотом) и вторым атомом углерода.
Енамины являются хорошими нуклеофилами и хорошими основаниями. Их поведение как углеродных нуклеофилов объясняется со ссылкой на следующие резонансные структуры.
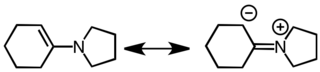
Формирование
Енамины являются лабильными и, следовательно, химически полезными фрагментами, которые можно легко получить из коммерчески доступных исходных реагентов. Обычный путь получения енамина - это катализируемая кислотой нуклеофильная реакция кетоновых (Stork, 1963) или альдегидных (Mannich / Davidsen, 1936) разновидностей, содержащих α-водород, со вторичными аминами. Кислотный катализ не всегда требуется, если pKa реагирующего амина достаточно высока (например, пирролидин, который имеет pKa 11,26). Если pKa реагирующего амин низкий, однако, тогда требуется кислотный катализ как на стадии добавления, так и на стадии дегидратации[7] (общие дегидратирующие агенты включают MgSO4 и Na2ТАК4).[8] Первичные амины обычно не используются для синтеза енаминов из-за преимущественного образования более термодинамически стабильных иминных форм.[9] Самоконденсация метилкетона - это побочная реакция, которой можно избежать, добавляя TiCl.4[10] в реакционную смесь (действовать как поглотитель воды).[11][12] Пример реакции альдегида со вторичным амином с образованием енамина через промежуточный карбиноламин показан ниже:
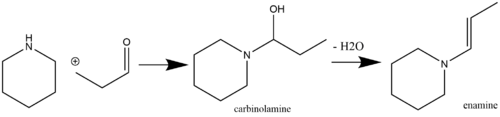
Реакции
Алкилирование
Хотя енамины более нуклеофильны, чем их енольные аналоги, они все же могут реагировать избирательно, что делает их пригодными для реакций алкилирования. Нуклеофил енамина может атаковать галогеналканы с образованием алкилированного иминий промежуточный солевой продукт, который затем гидролизуется с получением кетона (исходного материала в синтезе енамина). Эта реакция была впервые предложена Гилбертом Сторком, и ее иногда называют по имени ее изобретателя. Аналогично эту реакцию можно использовать как эффективное средство ацилирования. В этой реакции могут быть использованы различные алкилирующие и ацилирующие агенты, включая бензиловые, аллильные галогениды.[13]

Ацилирование
В реакции, очень похожей на алкилирование енаминов, енамины можно ацилировать с образованием конечного дикарбонильного продукта. Исходный енамин подвергается нуклеофильному присоединению к ацилгалогенидам с образованием промежуточной иминиевой соли, которая может гидролизоваться в присутствии кислоты.[14]

Металлоенамины
Сильные основания, такие как LiNR2, можно использовать для депротонирования иминов и образования металлоенаминов. Металлоенамины могут оказаться синтетически полезными из-за их нуклеофильности (они более нуклеофильны, чем еноляты). Таким образом, они лучше реагируют с более слабыми электрофилами (например, их можно использовать для открытия эпоксидов.[15]Наиболее заметно то, что эти реакции позволили осуществить асимметричное алкилирование кетонов посредством превращения в хиральные промежуточные металлоенамины.[16]
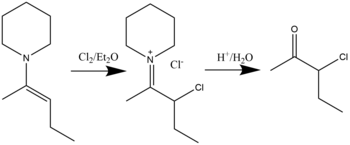
Галогенирование
Соединения β-галогениммония могут быть синтезированы посредством реакции енаминов с галогенидами в растворителе диэтиловом эфире. Гидролиз приведет к образованию α-галогенкетонов.[17] Было показано, что хлорирование, бромирование и даже йодирование возможно. Общая реакция показана ниже:
Окислительная связь
Енамины могут быть эффективно связаны с енольными силанами посредством обработки нитратом аммония Ce (IV). Об этих реакциях сообщила группа Нарасака в 1935 году, указав путь к стабильным енаминам, а также один пример 1,4-дикетона (полученного из реагента морфолинамина).[18] Позже эти результаты были использованы группой MacMillan при разработке органокатализатора, который использовал субстраты Narasaka для энантиоселективного получения 1,4-дикарбонилов с хорошими выходами.[19]Окислительная димеризация альдегидов в присутствии аминов протекает через образование енамина с последующим окончательным образованием пиррола.[20] Этот метод симметричного синтеза пиррола был разработан в 2010 году группой Jia как новый ценный способ синтеза пирролсодержащих природных продуктов.[21]
Аннулирование
Химия енаминов была реализована с целью получения энантиоселективной версии одного реактора Аннулирование Робинсона. В Аннулирование Робинсона, опубликованная Робертом Робинсоном в 1935 году, представляет собой реакцию, катализируемую основанием, которая объединяет кетон и метилвинилкетон (обычно сокращенно MVK) с образованием системы конденсированных колец с циклогексеноном. Эта реакция может быть катализатором пролин пройти через промежуточные хиральные енамины, которые обеспечивают хорошую стереоселективность.[22] Это важно, в частности, в области синтеза природных продуктов, например, для синтеза кетона Виланда-Мешера - жизненно важного строительного блока для более сложных биологически активных молекул.[23][24]
Реактивность
Енамины действуют как нуклеофилы, которым для реакционной способности требуется меньшая кислотно-щелочная активация, чем их енолятные аналоги. Также было показано, что они обладают большей селективностью и меньшим количеством побочных реакций. Существует градиент реакционной способности между различными типами енаминов, при этом кетоновые енамины обладают большей реакционной способностью, чем их альдегидные аналоги.[25]Циклические кетоновые енамины следуют тенденции реакционной способности, при которой пятичленное кольцо является наиболее реактивным из-за его максимально плоской конформации у азота, следуя тенденции 5> 8> 6> 7 (семичленное кольцо является наименее реакционно-способным). Эта тенденция была приписана количеству p-символа на орбитали неподеленной пары азота - более высокий p-символ, соответствующий большей нуклеофильности, потому что p-орбиталь допускает передачу на π-орбиталь алкена. Аналогично, если N неподеленная пара участвует в стереоэлектронных взаимодействиях с аминным фрагментом, неподеленная пара выскочит из плоскости (будет пирамидализироваться) и нарушит передачу в соседнюю π C-C связь.[26][27]

Есть много способов модулировать реакционную способность енамина в дополнение к изменению стерических / электронных компонентов в азотном центре, включая изменение температуры, растворителя, количества других реагентов и типа электрофила. Настройка этих параметров обеспечивает преимущественное образование енаминов E / Z, а также влияет на образование более / менее замещенного енамина из исходного материала кетона.[28]
Смотрите также
- Прекращает реакцию алкилирования гидразона SAMP / RAMP
- Реакция Хайоса – Пэрриша – Эдера – Зауэра – Вихерта.
- Майкл Аддишн
- Синтез индола Неницеску
- Органокатализ
- Аннулирование Робинсона
- Алкилирование енамином аиста
- Торп реакция
- Флуоксиместерон
Рекомендации
- ^ Клейден, Джонатан (2001). Органическая химия. Оксфорд, Оксфордшир: Издательство Оксфордского университета. ISBN 978-0-19-850346-0.
- ^ Смит, Майкл Б .; Марш, Джерри (2007), Продвинутая органическая химия: реакции, механизмы и структура (6-е изд.), Нью-Йорк: Wiley-Interscience, ISBN 978-0-471-72091-1
- ^ Enamines: Synthesis: Structure, and Reactions, Second Edition, Gilbert Cook (редактор). 1988, Марсель Деккер, Нью-Йорк. ISBN 0-8247-7764-6
- ^ Р. Б. Вудворд, И. Дж. Пахтер и М. Л. Шейнбаум (1974). «2,2- (Триметилендитио) циклогексанон». Органический синтез. 54: 39.CS1 maint: несколько имен: список авторов (связь); Коллективный объем, 5, п. 1014
- ^ Р. Д. Берпитт и Дж. Г. Твитт (1968). «Циклодеканон». Органический синтез. 48: 56.; Коллективный объем, 5, п. 277
- ^ Имины и енамины | PharmaXChange.info
- ^ Капон, Брайан; У, Чжэнь Пин (апрель 1990 г.). «Сравнение таутомеризации и гидролиза некоторых вторичных и третичных енаминов». Журнал органической химии. 55 (8): 2317–2324. Дои:10.1021 / jo00295a017.
- ^ Локнер, Джеймс. «Стехиометрическая химия енаминов» (PDF). Группа Баран, Исследовательский институт Скриппса. Получено 26 ноября 2014.
- ^ Фермер, Стивен (2013-10-16). «Реакции энамина». UC Davis Chem Wiki.
- ^ Карлсон, Р.; Нильссон, А (1984). «Улучшенная процедура синтеза тетрахлорида титана для синтеза енамина». Acta Chemica Scandinavica. 38B: 49–53. Дои:10.3891 / acta.chem.scand.38b-0049.
- ^ Локнер, Джеймс. «Стехиометрическая химия енаминов» (PDF). Группа Баран, Исследовательский институт Скриппса. Получено 26 ноября 2014.
- ^ Белый, Уильям Эндрю; Вайнгартен, Гарольд (январь 1967). «Универсальный синтез нового енамина». Журнал органической химии. 32 (1): 213–214. Дои:10.1021 / jo01277a052.
- ^ Уэйд, Л. (1999). Органическая химия. Река Сэдл, Нью-Джерси: Prentice Hall. стр.1019.
- ^ Фермер, Стивен (2013-10-16). «Реакции энамина». UC Davis Chem Wiki.
- ^ Эванс, Д. «Енолаты и металлоенамины II» (PDF). Получено 10 декабря 2014.[постоянная мертвая ссылка ]
- ^ Мейерс, А. И .; Уильямс, Дональд Р. (август 1978 г.). «Асимметричное алкилирование ациклических кетонов через хиральные металлоенамины. Влияние кинетических и термодинамических металлов». Журнал органической химии. 43 (16): 3245–3247. Дои:10.1021 / jo00410a034.
- ^ Зойферт, Уолтер; Эйфенбергер, Франц (1979). "Zur Halogenierung von Enaminen - Darstellung von β-Halogen-iminium-halogeniden". Chemische Berichte. 112 (5): 1670–1676. Дои:10.1002 / cber.19791120517.
- ^ Ито, Y; Konoike, T; Саегуса, Т. (1975). «Синтез 1,4-дикетонов реакцией силиленольного эфира с оксидом серебра. Региоспецифическое образование промежуточных продуктов енолята серебра (I)». Журнал Американского химического общества. 97 (3): 649–651. Дои:10.1021 / ja00836a034.
- ^ Jang, HY; Hong, JB; Макмиллан, DWC (2007). «Энантиоселективная органокаталитическая активация одиночных молекулярных орбиталей: энантиоселективная альфа-еноляция альдегидов» (PDF). Варенье. Chem. Soc. 129 (22): 7004–7005. Дои:10.1021 / ja0719428. PMID 17497866.
- ^ Ли, Q; Вентилятор, А; Lu, Z; Cui, Y; Линь, Вт; Цзя, Y (2010). «Однореакторный AgOAc-опосредованный синтез полизамещенных пирролов из первичных аминов и альдегидов: применение к полному синтезу пурпурона». Органические буквы. 12 (18): 4066–4069. Дои:10,1021 / ol101644g. PMID 20734981.
- ^ Го, Фэнхай; Клифт, Майкл Д .; Томсон, Риган Дж. (Сентябрь 2012 г.). «Окислительное связывание енолатов, енолсиланов и енаминов: методы и синтез природных продуктов». Европейский журнал органической химии. 2012 (26): 4881–4896. Дои:10.1002 / ejoc.201200665. ЧВК 3586739. PMID 23471479.
- ^ Список, Бенджамин (2002). «Асимметричные реакции, катализируемые пролином». Тетраэдр. 58 (28): 5573–5590. Дои:10.1016 / s0040-4020 (02) 00516-1.
- ^ Буй, Томми; Барбас (2000). «Асимметричное аннулирование Робинсона, катализируемое пролином». Буквы Тетраэдра. 41 (36): 6951–6954. Дои:10.1016 / s0040-4039 (00) 01180-1.
- ^ Винер, Джейк. «Энантиоселективный органический катализ: подходы, отличные от MacMillan» (PDF). Архивировано из оригинал (PDF) 26 октября 2017 г.. Получено 29 ноябрь 2014.
- ^ Хикмотт, Питер (май 1982). «Енамины: последние достижения в синтетических, спектроскопических, механистических и стереохимических аспектах - II». Тетраэдр. 38 (23): 3363–3446. Дои:10.1016/0040-4020(82)85027-8.
- ^ Майр, Х. (2003). «Связь структура-нуклеофильность для енаминов». Chem. Евро. J. 9 (10): 2209–18. Дои:10.1002 / chem.200204666. PMID 12772295.
- ^ Хикмотт, Питер (май 1982). «Енамины: последние достижения в синтетических, спектроскопических, механистических и стереохимических аспектах - II». Тетраэдр. 38 (23): 3363–3446. Дои:10.1016/0040-4020(82)85027-8.
- ^ Локнер, Джеймс. «Стехиометрическая химия енаминов» (PDF). Группа Баран, Исследовательский институт Скриппса. Получено 26 ноября 2014.