G & T-Seq - G&T-Seq - Wikipedia
G & T-seq (Короче для секвенирование одноклеточного генома и транскриптома) - это новая форма секвенирование одной клетки метод, позволяющий одновременно получить оба транскриптомный и геномный данные из отдельных клеток, что позволяет напрямую сравнивать данные экспрессии гена с соответствующими геномными данными в той же клетке ...[1]
Фон
Появление секвенирования отдельных клеток предоставило исследователям инструменты для решения генотипически и фенотипически отдельные клетки в смешанной популяции.[2] В случаях, когда такие неоднородность актуален, например, при опухолях, этот метод позволяет изучать клональные отношения и эволюция опухоли.[3] Кроме того, более детально можно изучить редкие типы клеток и образцы, содержащие небольшое количество клеток, например, в случае циркулирующих опухолевых клеток.[4] Однако предыдущие методы подготовка библиотеки обычно включает захват мРНК или геномной ДНК (гДНК), но не обоих одновременно.[5] Путем одновременного захвата и секвенирования ДНК и РНК с помощью метода, называемого секвенированием G&T, исследователи могут получать информацию о последовательности для анализа генома и транскриптома из библиотек отдельных клеток, что позволяет проводить интегрированные исследования с участием обеих сетей. В качестве доказательства концепции авторы G & T-seq продемонстрировали его способность приобретать как информационную РНК (мРНК), так и геномную ДНК (гДНК), используя парамагнитные бусины с биотинилированный олиго-дезокситиминовый (dT) праймер для отделения полиаденилированной (Поли-A) РНК от ее гДНК перед амплификацией и подготовкой библиотеки. Проверочные эксперименты по G & T-seq, проведенные с использованием клеточных линий с доступными ранее данными секвенирования, показывают, что секвенирование покрытия, профиль экспрессии гена, и Профили количества копий ДНК были надежно воспроизведены с помощью секвенирования G&T, и что этот метод смог вызвать большинство (87%) всех ранее аннотированных однонуклеотидных вариантов (SNV) в этих клеточных линиях. На этом основании авторы утверждали, что процесс физического отделения мРНК от гДНК не оказывает отрицательного влияния на выход или качество данных секвенирования.[1]
Методы
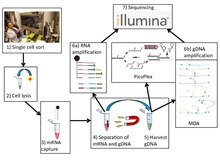
Подобно обычному секвенированию отдельных клеток, G & T-seq включает сбор и лизис желаемых клеток. Однако и гДНК, и полиА-мРНК захватываются и физически разделяются перед амплификацией и конструированием библиотеки для анализа с использованием платформы секвенирования.
Отделение полиаденилированной РНК от геномной ДНК
Секвенирование G&T отделяет мРНК от гДНК с использованием процедуры беспристрастной глобальной амплификации, описанной ранее.[6] Сначала мРНК выделяют на специализированном олиго-dT (5’-биотин-триэтиленгликоль-AAGCAGTGGTATCAACGCAGAGTAC (T) 30VN-3 ’), конъюгированном с парамагнитными бусами, связанными со стрептавидином.[7] Олиго-dT связывается с поли-A-хвостами процессированной мРНК, вылавливая их из пула геномного материала. Затем парамагнитные шарики пространственно изолированы намагничиванием. Геномный материал, оставшийся в супернатант извлекается и физически отделяется от мРНК.[1]
Амплификация и секвенирование
Авторы, разработавшие G & T-seq, использовали и подтвердили два метода амплификации всего генома: амплификацию с множественным смещением и PicoPlex. Другие методы, такие как МАЛЬБАК, может быть применимо, но еще не подтверждено.[1][8]
Множественное усиление смещения
Метод амплификации MDA можно использовать для генерации длинных высококачественных считываний, которые дают данные секвенирования сопоставимого качества с массовым секвенированием с использованием ПЦР-амплификация.[9] Этот метод включает использование гексамерных праймеров, которые случайным образом связываются с матрицей, с последующим удлинением ДНК с использованием ДНК-полимераза phi29. Достигнув 5’-конца расположенного ниже праймера, полимераза замещает эту удлиненную цепь для продолжения синтеза. Смещенная цепь становится открытой для спаривания с большим количеством праймеров, что позволяет амплифицировать смещенную цепь. Процесс продолжается и производит библиотеку разветвленной ДНК, которую можно разрезать и секвенировать. Авторы метода G&T обнаружили, что, хотя MDA, использованный в G&T-seq, давал геномный охват такой же широты, как MDA, выполняемый при обычном секвенировании отдельных клеток, распределение охвата считыванием было менее равномерным по геному.[1]
ПикоПлекс
Хотя MDA обеспечивает считывание более высокого качества, подходящее для анализа SNP, профили числа копий ДНК, полученные с помощью такого метода, не обладают высокой точностью и воспроизводимостью из-за его неравномерной амплификации.[5][10] Альтернативный метод под названием PicoPlex, разработанный Rubicon Genomics, показал лучшие результаты.[1] Здесь удлинение случайных праймеров, лигированных к адаптер создает дополнительный нить с адаптером, который при денатурировании и случайном повторном праймировании дает двухцепочечный фрагмент с дополнительными адаптерами. Денатурация в отдельные нити позволяет формировать петли для шпилек из-за комплементарной природы их адаптеров, создавая библиотеку петель шпильки, которую нельзя использовать для последующей амплификации, тем самым предотвращая экспоненциальное усиление начальной смещения.[11][12]
кДНК амплификация
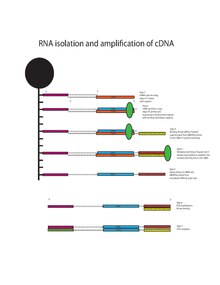
Информационная РНК, связанная с олиго-dT, обратно транскрибируется в кДНК с использованием праймеров oligo-dT с добавлением олигонуклеотида переключения шаблона (TSO, 5 "-AAGCAGTGGTATCAACGCAGAGTACrGrG + G-3’) и обратной транскриптазы Superscript II.[13][14] Обратная транскриптаза Superscript II обладает дополнительной концевой трансферазной активностью, которая добавляет различное количество остатков цитозина к концу 3’-концевой молекулы кДНК. Выступающие 3 ’остатки цитозина связываются с TSO, создавая расширенную матрицу. Обратная транскриптаза Superscript II переключает шаблоны и продолжает транскрипцию до завершения 3 ’конца кДНК. В результате получается полноразмерная кДНК, содержащая праймер 5 'oligo-dT, кДНК, транскрибируемую с мРНК, и универсальный сайт прайминга 3' для синтеза второй цепи. КДНК подвергается амплификации с использованием универсального праймера (5'-AAGCAGTGGTATCAACGCAGAGT-3 ' ) для 18 циклов ПЦР, прежде чем она подвергнется подготовке библиотеки с использованием набора Nextera XT Kit от Illumina и секвенированию с помощью платформы Illumina HiSeq.[1][15]
Альтернативные методы
Метод, аналогичный G & T-seq, разработанный несколькими месяцами ранее, - это DR-seq (секвенирование ДНК и РНК). Основное различие между двумя методами заключается в стадии амплификации, на которой амплификация ДНК и полиА-РНК происходит без их предварительного разделения.[16] DR-seq использует случайное праймирование, когда праймеры, содержащие общую 27-нуклеотидную последовательность вместе с вариабельной 8-нуклеотидной последовательностью (праймеры ad2), связываются с разными участками кДНК.[12] Несмотря на наличие нескольких (50-250) сайтов связывания праймеров на большинстве кДНК, каждый исходный (т.е. не продукт амплификации / in vitro) транскрипция ) Молекула кДНК обычно праймируется только один раз на начальном этапе амплификации, таким образом создавая один ампликон уникальной длины, содержащий праймер ad2 на 5 'конце. 3'-конец содержит праймер ad1, который является исходным праймером poly-dT, используемым для обратной транскрипции. Этот уникальный ампликон называется идентификатором на основе длины. Важно отметить, что идентификатор на основе длины создается, но не усиливается на этом этапе квазилинейной ПЦР. Затем количество уникальных идентификаторов на основе длины для каждого гена может быть использовано для определения количества исходных молекул кДНК (и, следовательно, мРНК), присутствующих для гена, что обеспечивает метод оценки экспрессии гена, позволяющий избежать эффекта смещения амплификации. Для дальнейшей амплификации кДНК для RNA-seq ампликоны кДНК, созданные на исходном этапе ПЦР, подвергаются транскрипции in vitro с использованием промотора Т7, включенного в праймер ad1, чтобы гарантировать, что транскрипты РНК происходят от кДНК, а не гДНК.
Преимущества метода DR-seq включают снижение вероятности контаминации и потери РНК, поскольку пропускается дополнительный этап разделения ДНК / РНК. Кроме того, смещение усиления снижается благодаря использованию вышеупомянутых идентификаторов на основе длины. Однако, поскольку ДНК и полиА-РНК не разделяются перед амплификацией и последующим секвенированием, экзонные области должны быть с вычислительной маской, оставляя только чтения, исходящие из гДНК, чтобы определить количество копий. Это создает проблемы для точного определения количества копий из гДНК. Авторы, тем не менее, отмечают, что на подсчет количества копий в больших областях генома явно не влияет маскирование, потому что кодирующие области составляют относительно небольшую часть генома.[16]
Приложения
Двойное секвенирование генома и транскриптома позволяет исследователям установить с высоким разрешением корреляции геномных аберраций с изменениями уровней транскрипции. Например, авторам этой методики удалось обнаружить единичные клетки с хромосомным анеуплоидии и установить, что эти анеуплоидии соответствовали повышенной или пониженной общей экспрессии хромосомных генов, когда имело место соответствующее хромосомное усиление (например, Трисомия ) или потеря. Субхромосомные изменения также могут быть коррелированы с изменениями экспрессии генов в пораженных локусах. Кроме того, авторам удалось найти транскрипт слияния и локализовать точку разрыва хромосомы в той же клетке, что привело к слиянию.[1]
G & T-seq также обеспечивает стратегию для установления причинных связей между генотипом и ассоциациями фенотипа в отдельных клетках (например, некодирующие SNV). Хотя массовое секвенирование генома и транскриптома может позволить связать набор генотипических признаков со средними паттернами экспрессии в популяции клеток, оно не учитывает тонкие или временные различия между отдельными клетками, которые могут возникать из-за экологии клетки.[17] Это представляет собой препятствие для исследователей, пытающихся точно определить геномные причины, лежащие в основе изменений транскриптов, особенно в сочетании с образцами опухолей, где широко распространена гетерогенность, а фоновая генетическая изменчивость может смешивать соответствующие мутации.[3][18][19] С другой стороны, обычное секвенирование отдельной клетки предотвращает прямую связь между мутациями и изменениями транскриптома, потому что при этом теряется либо ДНК, либо РНК. Традиционно исследователям приходилось использовать другие методы, такие как классификация на основе маркеров клеток. Однако такие методы дискриминации основаны на доступности специфических антител и обеспечивают относительно грубое различение по сравнению с секвенированием, поскольку экспрессия маркеров клеточной поверхности составляет лишь часть его общего фенотипа.[20][21]
Наконец, отделение ДНК от РНК открывает путь к двойному секвенированию эпигенома и транскриптома, двух компонентов клетки, которые неразрывно связаны друг с другом. Тем не менее, это потребует проверки с использованием обычной одиночной ячейки. бисульфитное секвенирование чтобы разделение ДНК и РНК не влияло на Метилирование ДНК положение дел.
Соображения
Смещение GC
Амплификация MDA имеет врожденную предвзятость в отношении повторяющихся последовательностей, которые недостаточно представлены в продуктах MDA. В контексте секвенирования G&T это приводит к уменьшенному количеству считываний как% от Содержимое GC увеличивается для конкретного региона.
Распределение охвата чтения
Сравнение амплификации одноклеточной остаточной геномной ДНК после выделения мРНК с помощью MDA с амплификацией геномной ДНК одной клетки без выделения мРНК с помощью MDA, показало менее равномерно распределенное покрытие по геному после выделения мРНК. Хотя наблюдалось сокращение распределения охвата, оно было незначительным.
Исключение альтернативной РНК
Выделение мРНК описанным методом G & T-seq способно улавливать только мРНК, которые имеют поли-A-хвост достаточной длины, который может быть захвачен приманкой oligo-dT.[6] Это не полное представление мРНК, присутствующей в клетке. Некоторые мРНК играют решающую роль в фенотипической экспрессии, но не имеют стандартной длины хвоста полиА из-за альтернативного полиаденилирования.[22] Следовательно, сравнение генотип-фенотип корреляции G&TS не обязательно представляет собой лучшую причинную связь между ними.
Корреляция экспрессии белка
Выделение мРНК - не единственное препятствие в установлении связи генотип-фенотип. Недостаточно использовать мРНК в качестве заменителя экспрессии общего белка, потому что существуют другие виды РНК, которые также играют важную роль в фенотипической экспрессии. Другой вспомогательный метод, который может поддержать утверждения, сделанные с помощью секвенирования G&T, - это полный протеомный анализ с помощью масс-спектрометрии, дающий лучшее представление о связи между геномными изменениями и фенотипическим представлением.[15]
Рекомендации
- ^ а б c d е ж грамм час Macaulay, I.C .; Haerty, W .; Kumar, P .; Li, Y. I .; Hu, T. X .; Teng, M. J .; Воет, Т. (2015). «G & T-seq: параллельное секвенирование одноклеточных геномов и транскриптомов». Методы природы. 12 (6): 519–22. Дои:10.1038 / nmeth.3370. PMID 25915121.
- ^ Ван, X. Секвенирование отдельных клеток и системная иммунология (Том 5). Springer
- ^ Си-Си Чен, Фань Бай (2015). «Одноклеточный анализ циркулирующих опухолевых клеток». 癌症 生物学 与 医学 : 文կ. 12 (3): 184–192. Дои:10.7497 / j.issn.2095-3941.2015.0056. ЧВК 4607822. PMID 26487963.
- ^ а б Grün, D .; ван Ауденаарден, А. (2015). «Дизайн и анализ экспериментов по секвенированию одной клетки». Клетка. 163 (4): 799–810. Дои:10.1016 / j.cell.2015.10.039. PMID 26544934.
- ^ а б Klein, C.A .; Seidl, S .; Petat-Dutter, K .; Offner, S .; Geigl, J. B .; Schmidt-Kittler, O .; Бауэрле П. А. (2002). «Комбинированный анализ транскриптома и генома отдельных микрометастатических клеток». Природа Биотехнологии. 20 (4): 387–92. Дои:10.1038 / nbt0402-387. PMID 11923846.
- ^ Picelli, S .; Faridani, O.R .; Björklund, Å. К .; Winberg, G .; Sagasser, S .; Сандберг, Р. (2014). «Полноразмерная последовательность РНК из отдельных клеток с использованием Smart-seq2». Протоколы природы. 9 (1): 171–81. Дои:10.1038 / nprot.2014.006. PMID 24385147.
- ^ Chapman, A. R .; Он, З .; Lu, S .; Yong, J .; Tan, L .; Tang, F .; Се, X.С. (2015). «Амплификация транскриптома одной клетки с MALBAC». PLOS ONE. 10 (3): e0120889. Дои:10.1371 / journal.pone.0120889. ЧВК 4378937. PMID 25822772.
- ^ Blanco, L .; Бернад, А .; Lázaro, J.M .; MartÃn, G .; Garmendia, C .; Салас, М. (1989). «Высокоэффективный синтез ДНК ДНК-полимеразой фага phi 29. Симметричный режим репликации ДНК». Журнал биологической химии. 264 (15): 8935–8940. PMID 2498321.
- ^ Voet, T .; Kumar, P .; Van Loo, P .; Cooke, S.L .; Marshall, J .; Lin, M .; Кэмпбелл, П. Дж. (2013). «Секвенирование одноклеточного парного генома выявляет структурные вариации в зависимости от клеточного цикла». Исследования нуклеиновых кислот. 41 (12): 6119–6138. Дои:10.1093 / nar / gkt345. ЧВК 3695511. PMID 23630320.
- ^ "PGS / PGD." Рубикон Геномика. N.p., н.о. Интернет. 25 февраля 2016 г.
- ^ а б Zong, C .; Lu, S .; Chapman, A. R .; Се, X.С. (2012). «Общегеномное обнаружение однонуклеотидных вариаций и вариаций числа копий одной клетки человека» (PDF). Наука. 338 (6114): 1622–1626. Дои:10.1126 / science.1229164. ЧВК 3600412. PMID 23258894.
- ^ Goetz, J. J .; Тримарчи, Дж. М. (2012). «Секвенирование транскриптома отдельных клеток с помощью Smart-Seq». Природа Биотехнологии. 30 (8): 763–765. Дои:10.1038 / nbt.2325. PMID 22871714.
- ^ «Набор для подготовки библиотеки ДНК Nextera XT». Подготовительный комплект для библиотеки ДНК Nextera XT. N.p., n.d. Интернет. 25 февраля 2016 г.
- ^ а б Maier, T .; Güell, M .; Серрано, Л. (2009). «Корреляция мРНК и белка в сложных биологических образцах». Письма FEBS. 583 (24): 3966–3973. Дои:10.1016 / j.febslet.2009.10.036. PMID 19850042.
- ^ а б Dey, S. S .; Kester, L .; Spanjaard, B .; Биенко, М .; ван Ауденаарден, А. (2015). «Комплексное секвенирование генома и транскриптома одной и той же клетки». Природа Биотехнологии. 33 (3): 285–289. Дои:10.1038 / nbt.3129. ЧВК 4374170. PMID 25599178.
- ^ Шапиро, Э .; Biezuner, T .; Линнарссон, С. (2013). «Технологии, основанные на секвенировании отдельных клеток, произведут революцию в науке о целом организме». Природа Обзоры Генетика. 14 (9): 618–630. Дои:10.1038 / nrg3542. PMID 23897237.
- ^ Сюй, X .; Hou, Y .; Инь, X .; Bao, L .; Tang, A .; Песня, Л .; Он, W. (2012). «Секвенирование одноклеточного экзома выявляет характеристики однонуклеотидной мутации опухоли почки». Клетка. 148 (5): 886–895. Дои:10.1016 / j.cell.2012.02.025. PMID 22385958.
- ^ Patel, A. P .; Тирош, И .; Trombetta, J. J .; Шалек, А.К .; Gillespie, S.M .; Wakimoto, H .; Луис, Д. Н. (2014). «Single-cell RNA-seq подчеркивает внутриопухолевую гетерогенность первичной глиобластомы». Наука. 344 (6190): 1396–1401. Дои:10.1126 / science.1254257. ЧВК 4123637. PMID 24925914.
- ^ Воан, Кристофер. «Новый способ сортировки клеток без ограничений традиционных методов». Центр новостей. Стэнфордская медицина, 30 марта 2015 г. Интернет. 25 февраля 2016 г.
- ^ Bidlingmaier, S .; Чжу, X .; Лю Б. (2008). «Применение и ограничения гликозилированных эпитопов CD133 человека в определении раковых стволовых клеток». Журнал молекулярной медицины. 86 (9): 1025–1032. Дои:10.1007 / s00109-008-0357-8. ЧВК 2585385. PMID 18535813.
- ^ De Klerk, E .; AC; Хоэн, П. (2015). «Альтернативная транскрипция, обработка и трансляция мРНК: выводы из секвенирования РНК». Тенденции в генетике. 31 (3): 128–139. Дои:10.1016 / j.tig.2015.01.001. PMID 25648499.