Руботтовое окисление - Rubottom oxidation
| Руботтовое окисление | |
|---|---|
| Названный в честь | Джордж М. Руботтом |
| Тип реакции | Органическая окислительно-восстановительная реакция |
| Идентификаторы | |
| Портал органической химии | руботон-окисление |
В Руботтовое окисление полезный, высокоурожайный химическая реакция между силиловые эфиры енола и пероксикислоты с получением соответствующего α-гидроксикарбонильного продукта.[1][2][3][4][5] Механизм реакции был предложен в своем первоначальном раскрытии А.Г. Бруком.[6][7] с дополнительными доказательствами, позже предоставленными Джорджем М. Руботтомом.[8] После Прилежаевское окисление простого силиленольного эфира с пероксикислотой с образованием силокси оксиран промежуточное раскрытие цикла, катализируемое кислотой, дает оксокарбений ион.[1][4] Затем этот промежуточный продукт участвует в миграции 1,4-силила (Перестановка ручья ) с получением α-силоксикарбонильного производного, которое можно легко превратить в α-гидроксикарбонильное соединение в присутствии кислоты, основания или источника фторида.[1][9][10]
Механизм реакции

История
В 1974 году три независимые группы сообщили о реакции, которая теперь известна как окисление Руботтома:[1] А.Г. Брук,[6] А. Хасснер,[11] и Г. Руботтом.[12] Значительный прецедент реакции уже существовал.[3] Например, еще в 1930-х годах было известно, что сильно енолизуемые β-дикарбонильные соединения будут реагировать с пероксикислотами, хотя на самом деле продуктом были α-гидрокси-β-дикарбонильные соединения только в 1950-х и 60-х годах.[13][14]
Значительная работа А.Г. Брука в 1950-х годах о механизмах миграции кремнийорганического соединения, которая теперь известна как Перестановки ручья.[15][16] В 1974 г. Хиткок описал озонолиз простых эфиров силил енола с образованием продукта карбоновой кислоты посредством окислительного расщепления, при котором миграции силила наблюдались как побочные реакции и исключительно в случае бициклической системы.[17]
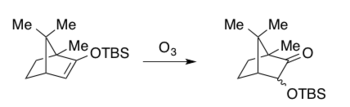
Общие особенности
Первоначальные реализации окисления Руботтома включали пероксикислоту мета-хлорпероксибензойная кислота (mCPBA) в качестве окислителя в дихлорметан (DCM) в случае Хасснера и Брука и гексаны для Руботтома.[6][11][12] Хотя с 1974 года реакция была изменена и модифицирована, mCPBA по-прежнему широко используется в качестве окислителя с немного большим разбросом в выборе растворителя.[1][4] DCM остается наиболее распространенным растворителем, за ним следуют различные углеводородные растворители, включая пентан и толуол.[1][4] Примечательно, что реакция протекает при относительно низких температурах, и нагревание выше комнатной температуры не требуется.[1][4] Низкие температуры позволяют регулировать стандартные условия окисления Руботтома с использованием множества чувствительных функций, что делает его идеальным для синтеза сложных молекул (см. Примеры синтеза ниже). Субстраты силиленольного эфира могут быть получены региоселективно из кетонов или альдегидов с применением термодинамического или кинетического контроля енолизации перед захватом желаемым источником кремнийорганического соединения (обычно хлоридом или трифлатом, например TBSCl или TBSOTf).[18] Как проиллюстрировано приведенными ниже примерами синтеза, простые эфиры силиленола могут быть выделены до воздействия условий реакции, или неочищенный материал может быть немедленно подвергнут окислению без выделения. Таким способом могут быть получены как ациклические, так и циклические производные силиленольного эфира, которые впоследствии могут быть использованы в качестве субстратов при окислении Руботтома.[1] Ниже приведены некоторые типичные продукты окисления Руботтома, синтезированные в основополагающих статьях.[6][11][12]

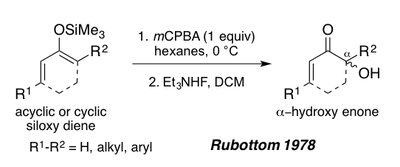
В 1978 году Руботтом показал, что силокси-1,3-диены, полученные из ациклических или циклических енонов, также могут служить субстратами для окисления Руботтома с целью образования α-гидроксиенонов после обработки фторидом триэтиламмония.[1][19] Эти субстраты дают единственный региоизомер в условиях реакции из-за богатой электронами природы пи-связи силил енола (см. Синтез перипланона B ниже).[1]
Модификации и улучшения
Окисление Rubottom осталось в значительной степени неизменным с момента его первоначального описания, но одним из основных недостатков стандартных условий является кислая среда, которая может привести к нежелательным побочным реакциям и разложению. Простая буферная система бикарбоната натрия обычно используется для решения этой проблемы, что особенно проблематично при синтезе бициклических и других сложных молекул (см. Примеры синтеза).[1][20] Введение хиральных окислителей также позволило синтезировать энантиочистые α-гидроксикарбонильные производные из их соответствующих силиленольных эфиров.[1] Первый пример энантиоселективное окисление Руботта был опубликован F.A. Davis[21] в 1987 году и продемонстрировал методологию хирального оксазиридина Дэвиса, дающую хорошие урожаи, но скромные энантиомерные избытки. В 1992 году К. Шарплесс показал, что асимметричное дигидроксилирование Условия, разработанные в его группе, можно было использовать для получения (R) - или (S) - α-гидроксикетонов из соответствующих силиленольных эфиров в зависимости от того, какие хиральные лиганды, полученные из алкалоидов чинхоны, были использованы.[22] Группы Ю. Ши[23] и В. Адам[24] опубликовали еще один энантиоселективный вариант окисления Руботтома в 1998 году с использованием хирального кетона Ши в присутствии оксона в буферной системе для получения α-гидроксикетонов с высоким выходом и высоким уровнем энантиомерный избыток. Группа Адама также опубликовала еще одну статью в 1998 году, в которой использовались комплексы марганца (III) - (Сален) в присутствии NaOCl (отбеливатель) в качестве окислителя и 4-фенилпиридин N-оксида в качестве добавки в системе с фосфатным буфером.[25] Эта методология также дает высокие выходы и энентиоселективность простых эфиров силиленола, а также ацеталей силилкетена, полученных из сложных эфиров.

Наряду с хиральными окислителями были изучены варианты mCPBA.[1] Станкович и Эспенсон опубликовали вариант окисления Руботтома, где метилтриоксорений используется в качестве каталитического окислителя в присутствии стехиометрических пероксид водорода.[1][26] Эта методология дает ациклические и циклические α-гидроксикетоны с высоким выходом с дешевым, коммерчески доступным окислителем. Неотъемлемой проблемой mCPBA является его неспособность окислять силилкетенацетали. Для синтеза α-гидроксиэфиров необходимы различные окислители, такие как NaOCl (см. Выше), ацетат свинца (IV) или комплекс гипофтористая кислота-ацетонитрил (HOF-ACN).[1][27] Группа Руботтома обнаружила, что ацетат свинца (IV) в DCM или бензоле дает хорошие выходы ациклических и циклических α-гидроксиэфиров после обработки неочищенной реакционной смеси фторидом триэтиламмония.[27] Позднее С. Розен использовал высокоэлектрофильный комплекс HOF-ACN для окисления различных электронно-богатых силиленольных эфиров, силилкетенацеталей и бис (силилацеталей), полученных из карбоновых кислот, с хорошими выходами при комнатной температуре или ниже. .[1][28]
Приложения в синтезе
Следующие ниже примеры представляют лишь небольшую часть синтезов, которые подчеркивают использование окисления Руботтома для установки важной α-гидрокси-функциональности. Некоторые из основных особенностей следующих синтезов включают использование буферных условий для защиты чувствительных субстратов и диастереоселективную установку α-гидроксигруппы из-за контролируемого субстратом смещения лица. Дополнительные примеры см. В справочниках.[1][3][4]
Окисление Руботтом было использовано в синтезе перипланон B, половой феромон, выделяемый самкой Американский таракан.[29][30] В синтезе использовался анионный окси-коп перегруппировка в сочетании с окислением Руботтома. После нагрева при наличии гидрид калия (KH) и 18-крон-6 (18-C-6) для воздействия на анионный окси-Cope промежуточный енолят улавливали триметилсилилхлоридом (TMSCl). Промежуточный силиленоловый эфир затем можно обработать mCPBA в условиях окисления Руботтома с получением желаемого α-гидроксикарбонильного соединения, которое затем может быть перенесено в (±) -перипланон B и его диастереомеры для подтверждения его структуры.

Бревизамид, предлагаемый биосинтетический предшественник морского токсина на основе простого полиэфира, был синтезирован Гошем и Ли, одной из стадий которого является окисление циклического силиленольного эфира по Руботту в буферных условиях.[31] Хиральный хромовый катализатор B был разработан Якобсен группы и придает высокие уровни энантио- и диастереоселективности.[32] Стереоцентры удобно расположить в Реакция Дильса-Альдера направляют окисление на менее затрудненную поверхность, давая единственный диастереомер, который затем может быть продолжен еще 14 стадиями до бревизамида.

Ван и его коллеги разработали надежный синтез мощного производного в килограммах. 2S-гидроксимутилин из плевромутилина, антибиотика, продуцируемого различными видами базидиомицеты.[33] Основным гидролизом для удаления фрагмента сложного гидроксилового эфира плевромутилина был получен мутилин. Последующее лечение гексаметилдисилазид лития (LiHMDS) и TMSCl давали силиленоловый эфир, защищенный TMS, который немедленно подвергали воздействию уксусная кислота - (HOAc) пиридин - (Py) забуференное окисление Руботтома перед кислотным гидролизом с получением 2S-гидроксимутилина. Эта оптимизированная последовательность имеет два важных аспекта. Во-первых, авторы первоначально генерировали простой эфир силил енола с использованием триэтиламина, который давал смесь желаемого кинетического продукта (показано ниже) нежелательного термодинамического продукта и гидролиза обратно до мутилина. Авторы обвинили образование кислотного побочного продукта триэтиламмония (pKa = 10,6) в нежелательных побочных продуктах и исправили это, используя LiHMDS исключительно для образования желаемого кинетического продукта без побочных реакций, катализируемых кислотой, из-за значительно более низкой кислотности протонированных продуктов. продукт (pKa = 26).[34] Во-вторых, в то время как окисление происходило с желаемой выпуклой стороны простого силиленольного эфира, авторы наблюдали значительное количество продуктов переокисления, которые они приписывали стабильности промежуточного оксокарбениевого иона в условиях, забуференных бикарбонатом натрия. Они выдвинули гипотезу, что увеличение продолжительности жизни промежуточных частиц приведет к чрезмерному окислению. После значительной оптимизации было обнаружено, что буфер HOAc / Py улавливает оксокарбениевое промежуточное соединение и предотвращает чрезмерное окисление с образованием исключительно 2S-гидроксимутилина после гидролиза силильных защитных групп.

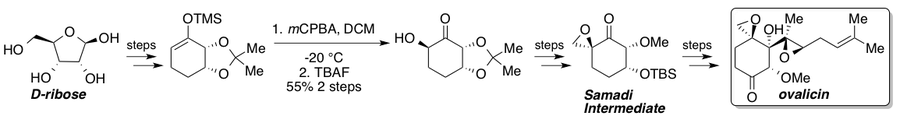
Овалицин, фумагиллин и их производные обладают сильными анти-ангиогенез свойства и видели многочисленные полные синтезы с момента их выделения.[35] Кори и Диттами сообщили о первом полном синтезе рацемического овалицина в 1985 г.[36] за которыми следуют два асимметричных синтеза, о которых в 1994 г. сообщил Самади[37][38] и Кори[39] который показал стратегию хирального пула от L-квебрахитол и асимметричное дигидроксилирование соответственно. В 2010 году Ядав и его коллеги сообщили о маршруте, который перехватил маршрут Самади из хирального пула исходного материала D-рибоза.[40] Стандартное окисление Руботтома дает единственный стереоизомер из-за субстратного контроля и представляет собой ключевой стереогенный этап на пути к кетону Самади. После синтеза кетон Самади может быть преобразован в (-) - овалицин с помощью известных стадий.
Велутинол А[41] был впервые синтезирован Исакой и сотрудниками.[42] Авторы показывают, что высокая региоселективность этой реакции направлена гидроксильной группой, синфазной с протоном слияния кольца. Реакции, в которых стереохимия гидроксильной группы инвертирована, приводили к снижению региоселективности, а удаление гидроксильной группы давало исключительное образование другого региоизомера. Вероятно, что непосредственная близость гидроксильной группы в син-изомере подкисляет протон слияния кольца за счет взаимодействия водородных связей, тем самым облегчая региоселективное депротонирование триэтиламином. Затем простой силиленоловый эфир обрабатывали избытком mCPBA для облегчения «двойного» окисления Руботтома с получением экзо-продукта с обеими гидроксильными группами вне конденсированной кольцевой системы. Затем этот дигидроксипродукт превращали в Велутинол А в три дополнительных этапа.

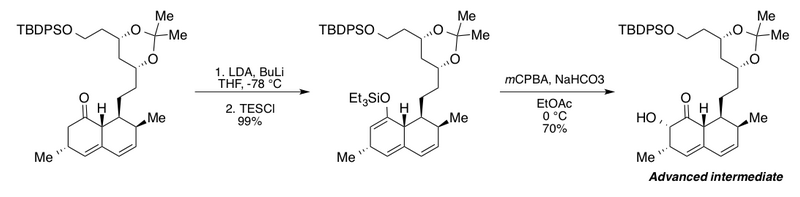
Группа Клайва использовала окисление Руботтома в синтезе продвинутого промежуточного продукта для своих исследований деградации холестерин -низкий метаболит грибов мевинолин.[2][43] В этой интересной последовательности добавлены лишние н-бутиллитий (BuLi) в присутствии диизопропиламид лития (LDA) для полного превращения производного бициклического кетона в соответствующий эфир силил енола. Авторы сообщают, что без BuLi максимальная доходность составляет всего 72%. Последующие буферные условия окисления Руботтома бикарбонатом натрия в этилацетате дали α-гидроксикетон в виде единственного диастереомера.
Группа Фальк синтезировала различные производные фосфатидил-D-мио-инозита, чтобы помочь в изучении различных фосфатидилинозитол-3-киназа (PI3K) клеточные сигнальные пути.[2][44] Их путь к сбору аналогов субстратов основан на контролируемом субстратом стереоселективном окислении Руботтома с использованием диметилдиоксиран (DMDO) как окислитель и каталитический камфорсульфоновая кислота (CSA), чтобы помочь в гидролизе. Относительно защитных групп см. Исх.[10]

Проблемы и недостатки
Хотя окисление Руботтом обычно дает хорошие выходы и хорошо масштабируется (см. Синтез 2S-гидроксимутилина), все еще существуют некоторые проблемы с реакцией. Как упоминалось выше, кислотные условия реакции не переносятся многими сложными субстратами, но это можно отменить с помощью буферных систем.[1] Плохая атомная экономия также является серьезной проблемой для реакции, поскольку для нее требуется стехиометрический окислитель, который приводит к образованию большого количества отходов.[3] С пероксидами также может быть опасно работать. Известно, что mCPBA детонирует от удара или искр.[45]
Хотя силиленольные эфиры альдегидов и кетонов являются традиционными субстратами для окисления Руботтома, как упоминалось выше, силилкетенацетали и бис (силилацетали) могут быть окислены до их α-гидроксиэфиров или производных карбоновой кислоты с использованием ацетата свинца (IV) или гипофтористая кислота -ацетонитрил (HOF – ACN).[27] Однако эти α-гидроксилирования не протекают через промежуточные соединения эфира силиленола и, следовательно, технически не являются окислениями Руботта. Для окисления многих из этих карбонильных производных после превращения их в соответствующий енолят или родственный анион можно использовать различные окислители. Некоторые распространенные окислители - это пероксикислоты, молекулярный кислород и реагенты гипервалентного йода.[5]
Рекомендации
- ^ а б c d е ж грамм час я j k л м п о п q р Kürti, стр. 388–389.
- ^ а б c Майерс, А.Г. Химия 215: Окисление. В архиве 2011-03-12 на Wayback Machine. chem.harvard.edu
- ^ а б c d Christoffers, J .; Baro, A .; Вернер, Т. (2004). «α-Гидроксилирование β-дикарбонильных соединений». Adv. Synth. Катал. 346 (23): 143–151. Дои:10.1002 / adsc.200303140.
- ^ а б c d е ж Ли, стр. 478–479.
- ^ а б Chen, B.C .; Чжоу, П .; Дэвис, Ф. А .; Циганек, Э. (2003) «α-Гидроксилирование енолатов и силилэнольных эфиров». Органические реакции; Эд. Оверман, Л. Wiley, Глава 1, стр. 1–355, Дои:10.1002 / 0471264180.or062.01.
- ^ а б c d Брук, А.Г .; Макрэ, Д. М. (1974). «1,4-Силильные перегруппировки силоксиалкенов в силоксикетоны во время перекисного окисления». J. Organomet. Chem. 77 (2): C19 – C21. Дои:10.1016 / S0022-328X (00) 81332-7.
- ^ Брук, А. Г. (1974). «Молекулярные перестройки кремнийорганических соединений». Соотв. Chem. Res. 7 (3): 77–84. Дои:10.1021 / ar50075a003.
- ^ Rubottom, G.M .; Gruber, J.M .; Бокман, Р. К., младший; Ramaiah, M .; Медвид, Дж. Б. (1978). «Уточнение механизма перегруппировки эпоксидов енольного силилового эфира». Tetrahedron Lett. 19 (47): 4603–4606. Дои:10.1016 / S0040-4039 (01) 85682-3.CS1 maint: несколько имен: список авторов (связь)
- ^ Майерс, А.Г. Химия 215: Защитные группы - защита гидроксильной группы на основе кремния. chem.harvard.edu
- ^ а б Коценски, П.Дж. (2005) Защита групп. 3-е издание, Thieme, стр. 188–230, ISBN 1588903761.
- ^ а б c Hassner, A .; Reuss, R.H .; Пинник, Х. В. (1975). «Синтетические методы. VIII. Гидроксилирование карбонильных соединений с помощью эфиров силил енола». J. Org. Chem. 40 (23): 3427–3429. Дои:10.1021 / jo00911a027.
- ^ а б c Rubottom, G.M .; Vazquez, M.A .; Пелегрина, Д. Р. (1974). «Надкислотное окисление эфиров триметилсилил енола: легкая процедура Α-гидроксилирования». Tetrahedron Lett. 15 (49–50): 4319–4322. Дои:10.1016 / S0040-4039 (01) 92153-7.
- ^ House, H.O .; Гэннон, В. Ф. (1958). «Реакция β-дикетонов с надкислотами». J. Org. Chem. 23 (6): 879–884. Дои:10.1021 / jo01100a030.
- ^ Hubert, A.J .; Старчер, П. С. (1968). «Окисление Байера-Виллигера алкилоксоциклогексанкарбоксилатов». J. Chem. Soc. C: 2500. Дои:10,1039 / j39680002500.
- ^ Kürti, стр. 64–65.
- ^ Ли, стр. 68–99.
- ^ Clark, R.D .; Хиткок, К. Х. Озонирование силилкетенов (1974). «Озонирование силилоксиалкенов». Tetrahedron Lett. 15 (23): 2027–2030. Дои:10.1016 / S0040-4039 (01) 82622-8.CS1 maint: несколько имен: список авторов (связь)
- ^ House, H.O .; Czuba, L.J .; Gall, M .; Олмстед, Х. Д. (1969). "Химия карбанионов. XVIII. Получение эфиров триметилсилил енола". J. Org. Chem. 34 (8): 2324–2336. Дои:10.1021 / jo01260a018.
- ^ Rubottom, G.M .; Грубер, Дж. М. (1978). "Окисление 2-триметилсилилокси-1,3-диенов М-хлорпербензойной кислотой. Синтез альфа-гидрокси- и альфа-ацетоксиэнонов". J. Org. Chem. 43 (8): 1599–1602. Дои:10.1021 / jo00402a030.
- ^ Jauch, J. Стереохимия окисления руботта бициклическими эфирами силил енола; Реакции синтеза и димеризации бициклических Α-гидроксикетонов (1994). «Стереохимия окисления руботта бициклическими силиленоловыми эфирами; реакции синтеза и димеризации бициклических α-гидроксикетонов». Тетраэдр. 50 (45): 12903–12912. Дои:10.1016 / S0040-4020 (01) 81209-6.CS1 maint: несколько имен: список авторов (связь)
- ^ Дэвис, Ф. А .; Шеппард А.С. (1987). «Окисление эфиров силил енола с использованием 2-сульфонилоксазиридинов. Синтез. -Силоксиэпоксидов и α-гидроксикарбонильных соединений». J. Org. Chem. 52 (5): 954–955. Дои:10.1021 / jo00381a051.
- ^ Hashiyama, T .; Morikawa, K .; Шарплесс, К. Б. (1992). «Альфа.-Гидроксикетоны с высокой энантиомерной чистотой от асимметричного дигидроксилирования эфиров енола». J. Org. Chem. 57 (19): 5067–5068. Дои:10.1021 / jo00045a011.
- ^ Zhu, Y .; Tu, Y .; Yu, H .; Ши, Ю. (1998). «Высокоэнантиоселективное эпоксидирование силиловых эфиров енола и сложных эфиров». Tetrahedron Lett. 39 (43): 7819–7822. Дои:10.1016 / S0040-4039 (98) 01711-0.
- ^ Adam, W .; Fell, R.T .; Saha-Möller, C.R .; Чжао, К.-Г. (1998). "Синтез оптически активных Α-гидроксикетонов путем энантиоселективного окисления эфиров силил енола диоксираном, производным фруктозы". Тетраэдр. 9 (3): 397–401. Дои:10.1016 / S0957-4166 (98) 00005-6.
- ^ Adam, W .; Fell, R.T .; Stegmann, V. R .; Саха-Мёллер, К. Р. (1998). «Синтез оптически активных Α-гидроксикарбонильных соединений путем каталитического, энантиоселективного окисления эфиров силил енола и кетеновых ацеталей с комплексами (сален) марганца (III)». Варенье. Chem. Soc. 120 (4): 708–714. Дои:10.1021 / ja9726668.
- ^ Станкович, С .; Эспенсон, Дж. Х. (1998). «Простое окисление эфиров силил енола перекисью водорода, катализируемое метилтриоксорением». J. Org. Chem. 63 (12): 4129–4130. Дои:10.1021 / jo972315b.
- ^ а б c Rubottom, G.M .; Gruber, J.M .; Marrero, R .; Жюв, Х. Д., младший; Ким, К. В. (1983). "Окисление алкилтриметилсилилкетеновых ацеталей карбоксилатами свинца (IV)". J. Org. Chem. 48 (25): 4940–4944. Дои:10.1021 / jo00173a031.CS1 maint: несколько имен: список авторов (связь)
- ^ Даян, С .; Bareket, Y .; Розен, С. (1999). «Эффективное Α-гидроксилирование карбонилов с использованием HOF • CH3Комплекс CN ». Тетраэдр. 55 (12): 3657–3664. Дои:10.1016 / S0040-4020 (98) 01173-9.
- ^ Тем не менее, W. C. (1979). «(±) -Перипланон-B. Полный синтез и структура полового возбудителя феромона американского таракана». Варенье. Chem. Soc. 101 (9): 2493–2495. Дои:10.1021 / ja00503a048.
- ^ Николау, К.С.; Соренсен, Э. Дж (1996) Классика в полном синтезе: цели, стратегии, методы, Wiley, стр. 211–220, ISBN 3527292314.
- ^ Ghosh, A.K .; Ли, Дж. (2009). «Асимметричный полный синтез бревизамида». Орг. Латыш. 11 (18): 4164–4167. Дои:10.1021 / ol901691d. ЧВК 2812931. PMID 19694486.
- ^ Dossetter, A. G .; Jamison, T. F .; Якобсен, Э. Н. (1999). «Высокоэнантио- и диастереоселективные реакции гетеро-Дильса-Альдера, катализируемые новыми хиральными тридентатными катализаторами хрома (III)». Энгью. Chem. Int. Эд. Англ.. 38 (16): 2398–2400. Дои:10.1002 / (SICI) 1521-3773 (19990816) 38:16 <2398 :: AID-ANIE2398> 3.0.CO; 2-E. PMID 10458800.
- ^ Wang, H .; Андемайкл, Ю. У .; Фогт, Ф. Г. (2009). «Масштабируемый синтез 2 S-гидроксимутилина с помощью модифицированного окисления руботта». J. Org. Chem. 74 (1): 478–481. Дои:10.1021 / jo801969e. PMID 19053581.
- ^ Эванс, Д.А. Chem 206: Таблица pKa В архиве 2013-10-02 в Wayback Machine. evans.harvard.edu
- ^ Yamaguchi, J .; Хаяси, Ю. (2010). «Синтезы фумагиллина и овалицина». Chem. Евро. J. 16 (13): 3884–3901. Дои:10.1002 / chem.200902433. PMID 20209516.
- ^ Кори, Э. Дж .; Диттами, Дж. П. (1985). «Полный синтез (+/-) - овалицина». Варенье. Chem. Soc. 107: 256–257. Дои:10.1021 / ja00287a049.
- ^ Ванна, с .; Биллингтон, Д. С.; Gero, S.D .; Quiclet-Sire, B .; Самади, М. (1994). «Полный синтез (-) - овалицина из L-кебрахитола». J. Chem. Soc. Chem. Commun. (12): 1495–1496. Дои:10.1039 / c39940001495.
- ^ Barton, D.H.R .; Ванна, с .; Биллингтон, Д. С .; Gero, S.D .; Quiclet-Sire, B.A .; Самади, М. (1995). «Полный синтез (-) - овалицина и аналогов из L-Quebrachitol». J. Chem. Soc., Perkin Trans. 1 (12): 1551–1558. Дои:10.1039 / п19950001551.
- ^ Кори, Э. Дж .; Guzman-Perez, A .; Ноэ М.С. (1994). «Короткий энантиоселективный синтез (-) - овалицина, мощного ингибитора ангиогенеза, с использованием субстрат-усиленного каталитического асимметричного дигидроксилирования». Варенье. Chem. Soc. 116 (26): 12109–12110. Дои:10.1021 / ja00105a084.
- ^ Yadav, J .; Reddy, P .; Редди, Б. (2010). «Стереоселективный тотальный синтез (-) - овалицина». Synlett. 2010 (3): 457–461. Дои:10.1055 / с-0029-1219191.
- ^ Yunes, R.A .; Pizzolatti, M. G .; Sant'Ana, A.E .; Hawkes, G.E .; Каликсто, Дж. Б. (1993). «Структура Велутинола, противовоспалительного соединения с новым скелетом беременной». Фитохимический анализ. 4 (2): 76–81. Дои:10.1002 / pca.2800040205.
- ^ Isaka, N .; Tamiya, M .; Hasegawa, A .; Исигуро, М. (2011). «Краткий полный синтез непептидного антагониста рецептора брадикинина B1 Велутинола А». Евро. J. Org. Chem. 2012 (4): 665–668. Дои:10.1002 / ejoc.201101728.
- ^ Клайв, Д. Л. Дж .; Чжан, К. (1995). «Исследования деградации мевинолина и компактина: формальный путь к полусинтетическим аналогам». J. Org. Chem. 60 (5): 1413–1427. Дои:10.1021 / jo00110a051.
- ^ Редди, К. К .; Саады, М .; Falck, J. R .; Уитед, Г. (1995). «Внутриклеточные медиаторы: синтез L-α-фосфатидил-D-мио-инозитол 3,4,5-трифосфата и аналогов глицерилового эфира». J. Org. Chem. 60 (11): 3385–3390. Дои:10.1021 / jo00116a023.
- ^ Технический бюллетень Sigma Aldrich mCPBA
Библиография
- Kürti, L .; Чако, Б. (2005) Стратегические применения названных реакций в органическом синтезе, Эльзевьер, ISBN 0124297854.
- Ли, Дж. Дж. (2009) Именные реакции: сборник подробных механизмов и синтетических приложений, 4-е издание, Springer, ISBN 8132204298