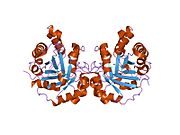
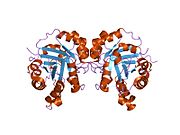
TPI1 - TPI1





Триозофосфат изомераза является фермент что у людей кодируется TPI1 ген.
Этот ген кодирует фермент, состоящий из двух идентичных белков, который катализирует изомеризацию глицеральдегид-3-фосфат (G3P) и дигидроксиацетонфосфат (DHAP) в гликолиз и глюконеогенез. Мутации в этом гене связаны с дефицитом триозофосфат-изомеразы. Псевдогены были идентифицированы на хромосомах 1, 4, 6 и 7. Альтернативная сварка приводит к множеству вариантов транскрипции.[5]
Структура
Триозофосфат-изомераза является членом альфа- и бета (α / β) классов белков; это гомодимер, и каждая субъединица содержит 247 аминокислот. Каждый мономер TPI1 содержит полный набор каталитических остатков, но фермент активен только в олигомерной форме.[6] Следовательно, фермент должен быть в димере для достижения полной функции фермента, даже если не предполагается, что два активных центра участвуют во взаимодействии друг с другом.[7] Каждая субъединица содержит 8 внешних альфа-спиралей, окружающих 8 внутренних бета-цепей, которые образуют консервативный структурный домен, называемый закрытым альфа / бета-стволом (αβ) или, более конкретно, ТИМ ствол. Характерной чертой большинства бочкообразных доменов TIM является присутствие активного сайта фермента в областях нижней петли, созданных восемью петлями, которые соединяют С-концы бета-цепей с N-концами альфа-спиралей. Бочковые белки TIM также имеют структурно консервативный фосфат связывающий мотив, с фосфатной группой, обнаруженной в субстрате или кофакторы.[5]
В каждой цепи неполярные аминокислоты, направленные внутрь от бета-цепей, вносят вклад в гидрофобное ядро структуры. Альфа-спирали амфипатичны: их внешние (контактирующие с водой) поверхности полярны, а их внутренние поверхности в значительной степени гидрофобны.
Функция
TPI катализирует перенос атома водорода от углерода 1 к углероду 2, внутримолекулярную реакцию окисления-восстановления. Эта изомеризация кетозы в альдозу протекает через СНГ-ендиол (ат) промежуточный. Эта изомеризация протекает без каких-либо кофакторов, и фермент дает 109 увеличение скорости по сравнению с неферментативной реакцией с участием химического основания (ацетат-ион).[8] Помимо своей роли в гликолизе, TPI также участвует в нескольких дополнительных метаболических биологических процессах, включая глюконеогенез, пентозофосфатный шунт и биосинтез жирных кислот.
Клиническое значение
Дефицит триозофосфатизомеразы - это заболевание, характеризующееся нехваткой эритроцитов (анемия), проблемами движения, повышенной восприимчивостью к инфекциям и мышечной слабостью, которое может влиять на дыхание и работу сердца. Анемия при этом состоянии начинается в младенчестве. Поскольку анемия возникает в результате преждевременного разрушения эритроцитов (гемолиза), она известна как гемолитическая анемия. Нехватка красных кровяных телец для переноса кислорода по всему телу приводит к сильной усталости (утомляемости), бледности кожи (бледности) и затрудненному дыханию. Когда красные клетки разрушаются, высвобождается железо и молекула, называемая билирубином; Люди с дефицитом триозофосфат-изомеразы имеют избыток этих веществ, циркулирующих в крови. Избыток билирубина в крови вызывает желтуху, то есть пожелтение кожи и белков глаз. Проблемы с движением обычно становятся очевидными к 2 годам у людей с дефицитом триозофосфат-изомеразы. Проблемы с движением вызваны повреждением двигательных нейронов, которые представляют собой специализированные нервные клетки в головном и спинном мозге, контролирующие движение мышц. Это нарушение приводит к мышечной слабости и истощению (атрофии) и вызывает проблемы с движением, типичные для дефицита триозофосфатизомеразы, включая непроизвольное напряжение мышц (дистония), тремор и слабый мышечный тонус (гипотония). У больных также могут развиться судороги. Слабость других мышц, таких как сердце (состояние, известное как кардиомиопатия) и мышца, отделяющая брюшную полость от грудной полости (диафрагма), также может возникать при дефиците триозофосфатизомеразы. Слабость диафрагмы может вызвать проблемы с дыханием и в конечном итоге приводит к дыхательной недостаточности. Лица с дефицитом триозофосфатизомеразы подвержены повышенному риску развития инфекций, потому что у них плохо функционируют белые кровяные клетки. Эти клетки иммунной системы обычно распознают и атакуют чужеродных захватчиков, таких как вирусы и бактерии, для предотвращения инфекции. Наиболее частыми инфекциями у людей с дефицитом триозофосфат-изомеразы являются бактериальные инфекции дыхательных путей. Люди с дефицитом триозофосфатизомеразы часто не доживают до детства из-за дыхательной недостаточности. В нескольких редких случаях пораженные люди без серьезного повреждения нервов или мышечной слабости доживают до взрослого возраста.[5] Дефицит чаще всего вызывается мутациями в TPI1, хотя были идентифицированы мутации в других изоформах. Обычным маркером дефицита TPI является повышенное накопление DHAP в экстрактах эритроцитов; это связано с тем, что дефектный фермент больше не может катализировать изомеризацию до GAP. Точечная мутация не влияет на скорость катализа, а скорее влияет на сборку фермента в гомодимер.[9][10]
Недавние открытия в Болезнь Альцгеймера исследования показали, что амилоид бета пептид-индуцированное нитроокислительное повреждение способствует нитротирозированию TPI в клетках нейробластомы человека.[11] Было обнаружено, что нитрозилированный TPI присутствует в препаратах мозга двойных трансгенных мышей, сверхэкспрессирующих человеческий белок-предшественник амилоида, а также у пациентов с болезнью Альцгеймера. В частности, нитротирозинирование происходит на Tyr164 и Tyr208 внутри белка, которые находятся недалеко от центра катализа; эта модификация коррелирует со сниженной активностью изомеризации.
Интерактивная карта проезда
Нажмите на гены, белки и метаболиты ниже, чтобы ссылки на соответствующие статьи.[§ 1]

- ^ Интерактивную карту путей можно редактировать на WikiPathways: «ГликолизГлюконеогенез_WP534».
Модельные организмы
| Характеристика | Фенотип |
|---|---|
| Гомозигота жизнеспособность | Аномальный |
| Рецессивный смертельное исследование | Аномальный |
| Плодородие | Нормальный |
| Масса тела | Нормальный |
| Беспокойство | Нормальный |
| Неврологический осмотр | Нормальный |
| Сила захвата | Нормальный |
| Горячая тарелка | Нормальный |
| Дисморфология | Нормальный |
| Косвенная калориметрия | Нормальный |
| Тест толерантности к глюкозе | Нормальный |
| Слуховой ответ ствола мозга | Нормальный |
| DEXA | Нормальный |
| Рентгенография | Нормальный |
| Температура тела | Нормальный |
| Морфология глаза | Нормальный |
| Клиническая химия | Нормальный |
| Плазма иммуноглобулины | Нормальный |
| Гематология | Нормальный |
| Лимфоциты периферической крови | Нормальный |
| Микроядерный тест | Нормальный |
| Вес сердца | Нормальный |
| Гистопатология кожи | Нормальный |
| Гистопатология головного мозга | Нормальный |
| Сальмонелла инфекционное заболевание | Аномальный[12] |
| Citrobacter инфекционное заболевание | Нормальный[13] |
| Все тесты и анализы от[14][15] |
Модельные организмы были использованы при изучении функции TPI1. Условный нокаутирующая мышь линия, называемая Tpi1tm1a (EUCOMM) Wtsi[16][17] был создан как часть Международный консорциум Knockout Mouse программа - проект по мутагенезу с высокой пропускной способностью для создания и распространения моделей болезней на животных среди заинтересованных ученых.[18][19][20]
Самцы и самки животных прошли стандартизованный фенотипический скрининг для определения последствий удаления.[14][21] Было проведено 26 испытаний мутант мышей и три значительных отклонения от нормы.[14] Нет гомозиготный мутант эмбрионы были идентифицированы во время беременности, и поэтому ни один из них не выжил до отлучение от груди. Остальные испытания проводились на гетерозиготный мутантные взрослые мыши и повышенная восприимчивость к бактериальная инфекция наблюдалась у животных-самцов.[14]
Смотрите также
Рекомендации
- ^ а б c ГРЧ38: Ансамбль выпуск 89: ENSG00000111669 - Ансамбль, Май 2017
- ^ а б c GRCm38: выпуск Ensembl 89: ENSMUSG00000023456 - Ансамбль, Май 2017
- ^ "Справочник человека по PubMed:". Национальный центр биотехнологической информации, Национальная медицинская библиотека США.
- ^ "Ссылка на Mouse PubMed:". Национальный центр биотехнологической информации, Национальная медицинская библиотека США.
- ^ а б c «Энтрез Ген: триозофосфатизомераза 1 TPI1».
- ^ Родригес-Алмазан С., Арреола Р., Родригес-Ларреа Д., Агирре-Лопес Б., де Гомес-Пуйу М. Т., Перес-Монфор Р., Костас М., Гомес-Пуйу А., Торрес-Лариос А. (август 2008 г.). «Структурная основа дефицита триозофосфатизомеразы человека: мутация E104D связана с изменениями консервативной водной сети на границе раздела димеров». Журнал биологической химии. 283 (34): 23254–63. Дои:10.1074 / jbc.M802145200. PMID 18562316.
- ^ Schnackerz KD, Gracy RW (июль 1991 г.). «Исследование каталитических центров триозофосфатизомеразы с помощью 31P-ЯМР с обратимо и необратимо связывающимися аналогами субстрата». Европейский журнал биохимии / FEBS. 199 (1): 231–8. Дои:10.1111 / j.1432-1033.1991.tb16114.x. PMID 2065677.
- ^ Давенпорт Р.К., Баш ПА, Ситон Б.А., Карплюс М., Петско Г.А., Риндж Д. (июнь 1991 г.). «Структура комплекса триозофосфат-изомераза-фосфогликологидроксамат: аналог промежуточного соединения на пути реакции». Биохимия. 30 (24): 5821–6. Дои:10.1021 / bi00238a002. PMID 2043623.
- ^ Ralser M, Heeren G, Breitenbach M, Lehrach H, Krobitsch S (20 декабря 2006 г.). «Дефицит триозофосфатизомеразы вызван измененной димеризацией, а не каталитической неактивностью мутантных ферментов». PLOS ONE. 1: e30. Дои:10.1371 / journal.pone.0000030. ЧВК 1762313. PMID 17183658.
- ^ Шнайдер А.С. (март 2000 г.). «Дефицит триозофосфатизомеразы: исторические перспективы и молекулярные аспекты». Лучшие практики и исследования Байера. Клиническая гематология. 13 (1): 119–40. Дои:10.1053 / бэха.2000.0061. PMID 10916682.
- ^ Guix FX, Ill-Raga G, Bravo R, Nakaya T, de Fabritiis G, Coma M, Miscione GP, Villà-Freixa J, Suzuki T, Fernàndez-Busquets X, Valverde MA, de Strooper B, Muñoz FJ (май 2009 г.) . «Амилоид-зависимое нитротирозинирование триозофосфатизомеразы вызывает гликирование и фибрилляцию тау». Мозг. 132 (Pt 5): 1335–45. Дои:10.1093 / мозг / awp023. PMID 19251756.
- ^ "Сальмонелла данные о заражении для Tpi1 ". Wellcome Trust Институт Сэнгера.
- ^ "Citrobacter данные о заражении для Tpi1 ". Wellcome Trust Институт Сэнгера.
- ^ а б c d Гердин А.К. (2010). "Программа генетики Sanger Mouse: характеристика мышей с высокой пропускной способностью". Acta Ophthalmologica. 88: 925–7. Дои:10.1111 / j.1755-3768.2010.4142.x. S2CID 85911512.
- ^ Портал ресурсов мыши, Институт Wellcome Trust Sanger.
- ^ «Международный консорциум нокаут-мышей».
- ^ "Информатика генома мыши".
- ^ Скарнес В.К., Розен Б., Вест А.П., Кутсуракис М., Бушелл В., Айер В., Мухика А.О., Томас М., Харроу Дж., Кокс Т., Джексон Д., Северин Дж., Биггс П., Фу Дж., Нефедов М., де Йонг П.Дж., Стюарт AF, Брэдли А. (июнь 2011 г.). «Ресурс условного нокаута для полногеномного исследования функции генов мыши». Природа. 474 (7351): 337–42. Дои:10.1038 / природа10163. ЧВК 3572410. PMID 21677750.
- ^ Долгин Э (июнь 2011 г.). "Библиотека мыши настроена на нокаут". Природа. 474 (7351): 262–3. Дои:10.1038 / 474262a. PMID 21677718.
- ^ Коллинз Ф.С., Россант Дж., Вурст В. (январь 2007 г.). «Мышь по всем причинам». Клетка. 128 (1): 9–13. Дои:10.1016 / j.cell.2006.12.018. PMID 17218247. S2CID 18872015.
- ^ ван дер Вейден Л., Уайт Дж. К., Адамс Д. Д., Логан Д. В. (2011). «Набор инструментов генетики мышей: раскрытие функции и механизма». Геномная биология. 12 (6): 224. Дои:10.1186 / gb-2011-12-6-224. ЧВК 3218837. PMID 21722353.
дальнейшее чтение
- Атиону А., Хамфрис А. (декабрь 1998 г.). «Возможность заместительной терапии наследственного нарушения гликолиза: дефицит триозофосфатизомеразы (обзор)». Международный журнал молекулярной медицины. 2 (6): 701–4. Дои:10.3892 / ijmm.2.6.701. PMID 9850739.
- Олах Дж., Орос Ф., Кесеру Г.М., Ковари З., Ковач Дж., Холлан С., Овади Дж. (Апрель 2002 г.). «Дефицит триозофосфатизомеразы: нейродегенеративная болезнь неправильной укладки». Сделки Биохимического Общества. 30 (2): 30–8. Дои:10.1042 / BST0300030. PMID 12023819.
- Rethoré MO, Kaplan JC, Junien C, Lejeune J (апрель 1977 г.). «12pter к 12p12.2: возможное отнесение триозофосфатизомеразы человека». Генетика человека. 36 (2): 235–7. Дои:10.1007 / BF00273263. PMID 858628. S2CID 25241150.
- Perry BA, Mohrenweiser HW (март 1992 г.). «Человеческая триозофосфатизомераза: замена Arg на Gly в положении 122 в термолабильном электроморфе, TPI-Manchester». Генетика человека. 88 (6): 634–8. Дои:10.1007 / BF02265287. PMID 1339398. S2CID 35721080.
- Доусон С.Дж., Белый Лос-Анджелес (май 1992 г.). «Лечение эндокардита Haemophilus aphrophilus ципрофлоксацином». Журнал инфекции. 24 (3): 317–20. Дои:10.1016 / S0163-4453 (05) 80037-4. PMID 1602151.
- Boyer TG, Maquat LE (ноябрь 1990 г.). «Минимальные требования к последовательности и факторам для инициации транскрипции с атипичного, содержащего TATATAA промотора домашнего хозяйства». Журнал биологической химии. 265 (33): 20524–32. PMID 2243103.
- Maquat LE, Chilcote R, Ryan PM (март 1985 г.). «Человеческая кДНК триозофосфатизомеразы и структура белка. Исследования дефицита триозофосфатизомеразы у человека». Журнал биологической химии. 260 (6): 3748–53. PMID 2579079.
- Даар И.О., Артимюк П.Дж., Филипс, округ Колумбия, Макват Л.Е. (октябрь 1986 г.). «Дефицит триозофосфатизомеразы человека: замена одной аминокислоты приводит к образованию термолабильного фермента». Труды Национальной академии наук Соединенных Штатов Америки. 83 (20): 7903–7. Дои:10.1073 / пнас.83.20.7903. ЧВК 386831. PMID 2876430.
- Бойер Т.Г., Круг-младший, Макват Л.Е. (март 1989 г.). «Транскрипционные регуляторные последовательности гена домашнего хозяйства триозофосфатизомеразы человека». Журнал биологической химии. 264 (9): 5177–87. PMID 2925688.
- Браун-младший, Даар И.О., Круг-младший, Макват Л.Е. (июль 1985 г.). «Характеристика функционального гена и нескольких процессированных псевдогенов в семействе генов триозофосфатизомеразы человека». Молекулярная и клеточная биология. 5 (7): 1694–706. Дои:10.1128 / mcb.5.7.1694. ЧВК 367288. PMID 4022011.
- Лу Х.С., Юань П.М., Грейси Р.В. (октябрь 1984 г.). «Первичная структура триозофосфатизомеразы человека». Журнал биологической химии. 259 (19): 11958–68. PMID 6434534.
- Mande SC, Mainfroid V, Kalk KH, Goraj K, Martial JA, Hol WG (май 1994). «Кристаллическая структура рекомбинантной триозофосфат-изомеразы человека с разрешением 2,8 A. Генетические нарушения человека, связанные с триозофосфат-изомеразой, и сравнение с трипаносомальным ферментом». Белковая наука. 3 (5): 810–21. Дои:10.1002 / pro.5560030510. ЧВК 2142725. PMID 8061610.
- Маруяма К., Сугано С. (январь 1994 г.). «Олиго-кэппинг: простой метод замены кэп-структуры эукариотических мРНК олигорибонуклеотидами». Ген. 138 (1–2): 171–4. Дои:10.1016/0378-1119(94)90802-8. PMID 8125298.
- Чанг М.Л., Артимюк П.Дж., Ву X, Холлан С., Ламми А., Макват Л.Е. (июнь 1993 г.). «Дефицит триозофосфатизомеразы человека в результате мутации Phe-240». Американский журнал генетики человека. 52 (6): 1260–9. ЧВК 1682273. PMID 8503454.
- Watanabe M, Zingg BC, Mohrenweiser HW (февраль 1996 г.). «Молекулярный анализ ряда аллелей у людей с пониженной активностью в локусе триозофосфатизомеразы». Американский журнал генетики человека. 58 (2): 308–16. ЧВК 1914533. PMID 8571957.
- Mainfroid V, Terpstra P, Beauregard M, Frère JM, Mande SC, Hol WG, Martial JA, Goraj K (март 1996). «Три мутанта hTIM, которые по-новому понимают, почему TIM является димером». Журнал молекулярной биологии. 257 (2): 441–56. Дои:10.1006 / jmbi.1996.0174. PMID 8609635.
- Ансари-Лари М.А., Музны Д.М., Лу Дж., Лу Ф., Лилли С.Э., Спанос С., Малли Т., Гиббс Р.А. (апрель 1996 г.). «Богатый генами кластер между генами CD4 и триозофосфат-изомеразы на хромосоме 12p13 человека». Геномные исследования. 6 (4): 314–26. Дои:10.1101 / гр.6.4.314. PMID 8723724.
- Ансари-Лари М.А., Шен Й., Музны Д.М., Ли В., Гиббс Р.А. (март 1997 г.). «Крупномасштабное секвенирование хромосомы 12p13 человека: экспериментальное и расчетное определение структуры гена». Геномные исследования. 7 (3): 268–80. Дои:10.1101 / гр.7.3.268. PMID 9074930.
- Расмуссен Р.К., Джи Х., Эддес Дж.С., Мориц Р.Л., Рид Г.Е., Симпсон Р.Дж., Доров Д.С. (1997). «Двумерный электрофоретический анализ белков карциномы молочной железы человека: картирование белков, которые связываются с доменом SH3 киназы смешанного происхождения MLK2». Электрофорез. 18 (3–4): 588–98. Дои:10.1002 / elps.1150180342. PMID 9150946. S2CID 37336552.
- Джи Х., Рид Г.Е., Мориц Р.Л., Эддес Дж.С., Берджесс А.В., Симпсон Р.Дж. (1997). «Двумерная база данных гелей белков карциномы толстой кишки человека». Электрофорез. 18 (3–4): 605–13. Дои:10.1002 / elps.1150180344. PMID 9150948. S2CID 25454450.
внешняя ссылка
- Обзор всей структурной информации, доступной в PDB за UniProt: P60174 (Триозофосфатизомераза) на PDBe-KB.
PDB галерея | |
|---|---|
|