Наследственная диффузная лейкоэнцефалопатия со сфероидами - Hereditary diffuse leukoencephalopathy with spheroids
| Наследственная диффузная лейкоэнцефалопатия со сфероидами (ЛПВП) | |
|---|---|
| Другие имена | Лейкоэнцефалопатия с началом у взрослых с аксональными сфероидами и пигментированной глией, аутосомно-доминантная лейкоэнцефалопатия с нейроаксональными сфероидами |
 | |
| Наследственная диффузная лейкоэнцефалопатия со сфероидами наследуется по аутосомно-доминантному типу. | |
Наследственная диффузная лейкоэнцефалопатия со сфероидами (HDLS) - редко встречающееся у взрослых аутосомно-доминантное заболевание был характеризован мозговой белое вещество дегенерация с демиелинизацией и аксональные сфероиды что приводит к прогрессирующей когнитивной и двигательной дисфункции. Сфероиды - это вздутие аксонов с прерывистым или отсутствующим миелин ножны. Считается, что заболевание возникает из-за первичной дисфункции микроглии, которая приводит к вторичному нарушению целостности аксонов, нейроаксональному повреждению и фокальным аксональным сфероидам, приводящим к демиелинизация. Сфероиды в HDLS в некоторой степени напоминают те, которые производятся напряжение сдвига в закрытая травма головы с повреждением аксонов, вызывая их набухание из-за блокировки аксоплазматический транспорт. Помимо травмы, аксональные сфероиды могут быть обнаружены в пожилом мозге, инсульте и других дегенеративных заболеваниях.[1] При HDLS неясно, происходит ли демиелинизация до аксональных сфероидов или что вызывает нейродегенерацию после явно нормального развития мозга и белого вещества, хотя генетический дефицит предполагает, что демиелинизация и аксональная патология могут быть вторичными по отношению к дисфункции микроглии.[2] Клинический синдром у пациентов с ЛПВП неспецифичен, и его можно принять за Болезнь Альцгеймера, лобно-височная деменция, атипичный паркинсонизм, рассеянный склероз, или же кортикобазальная дегенерация.[3]
Симптомы
При симптомах изменения личности, изменения поведения, слабоумие, депрессия и эпилепсии, ЛПВП часто ошибочно диагностируют при ряде других заболеваний.[4] Например, деменция или лобно-височные изменения поведения обычно заставляли некоторых клиницистов ошибочно принимать во внимание такие диагнозы, как болезнь Альцгеймера, лобно-височная деменция или атипичный паркинсонизм. Наличие изменений белого вещества привело к ошибочной диагностике рассеянного склероза. HDLS обычно проявляется психоневрологический симптомы прогрессируют до слабоумия, а через несколько лет проявляются двигательные нарушения. Со временем пациенты становятся прикованными к инвалидной коляске или прикованными к постели.[3]
Дегенерация белого вещества связана с другими лейкодистрофиями у взрослых, такими как метахроматическая лейкодистрофия (MLD), Болезнь Краббе (лейкодистрофия глобоидных клеток) и Х-сцепленный адренолейкодистрофия (X-ADL).[2]
| Болезнь | Эксклюзивная черта |
|---|---|
| MLD | Накопление метахроматического материала в белом веществе |
| Болезнь Краббе | Наличие глобоидных клеток, происходящих из микроглии, которые имеют несколько ядер. |
| X-ALD | Преобладающая патология теменно-затылочного белого вещества |
| Болезнь исчезающего белого вещества (VWM) |
|
| Насу-Хакола |
|
Психоневрологические симптомы
Многие нейропсихиатрические симптомы были выявлены в клинических исследованиях пациентов с ЛПВП. К ним относятся тяжелая депрессия и тревога, которые были выявлены примерно в 70% семей с ЛПВП, граничащие с суицидными наклонностями и злоупотребление алкоголем или наркотиками Такие как алкоголизм. Кроме того, пациенты могут проявлять дезориентацию, замешательство, возбуждение, раздражительность, агрессивность, измененное психическое состояние, потерю способности выполнять выученные движения (апраксия ) или неспособность говорить (мутизм ).[3]
Нарушение моторики
Люди с ЛПВП могут страдать от тремора, снижения подвижности тела, неустойчивости (Паркинсонизм, мышцы на одной стороне тела в постоянном сокращении (спастический гемипарез ), нарушение двигательной и сенсорной функции нижних конечностей (парапарез ), паралич с частичной или полной потерей всех конечностей и туловища (тетрапарез ), а также отсутствие произвольной координации движений мышц (атаксия ).[3]
Причины
Причиной ЛПВП в большинстве семей является мутация в рецептор колониестимулирующего фактора 1 (CSF1R), фактор роста микроглии и моноцитов / макрофагов, что позволяет предположить, что дисфункция микроглии может быть первичной при ЛПВП.[4]
Мутации сосредоточены в тирозин киназа домен (TKD) белка. Мутации в основном были обнаружены в экзонах 12-22 внутриклеточный ТКД, в том числе 10 миссенс-мутации у которых есть сингл нуклеотид делеция и делеция одного кодона, который состоит из триплета нуклеотидов, которые были удалены, что привело к целому аминокислота не кодироваться. Кроме того, три мутации сайта сплайсинга были выявлены, что вызвало удаление в кадре из экзон, выраженная нуклеотидная последовательность, приводящая к удалению более 40 аминокислот в TKD.[4]
Это определение основано на генетических исследованиях 14 семейств ЛПВП, подтверждающих мутации в этом гене. Белок рецептора CSF1 в основном участвует в регуляции, выживании, пролиферации и дифференцировке клеток микроглии.[5] Механизм дисфункции микроглии из-за мутаций в CSF1R, приводящих к потере миелина и образованию аксонального сфероида, остается неизвестным. Чтобы лучше понять болезнь, необходимы дальнейшие исследования. патогенез.
Патология
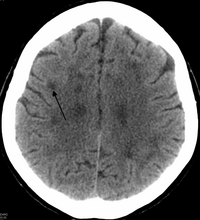
При HDLS наблюдается увеличение боковые желудочки и заметное истончение или ослабление белого вещества головного мозга.[6] Потеря белого вещества вызвана миелин потеря. Эти изменения связаны с диффузным глиоз умеренная потеря аксоны и многие аксональные сфероиды.[1]
Активированная или амебоидная микроглия и макрофаги которые содержат обломки миелина, липидные капли и коричневые гранулы автофлуоресцентного пигмента обнаруживаются в областях с демиелинизацией и аксональными сфероидами. В сильно дегенерированных областях много больших реактивных астроцитов, заполненных глиальным фибриллы.[1]
В случаях вскрытия было показано, что аномалии белого вещества относительно ограничены головной мозг избегая при этом мозжечок и многие из основных волоконных трактов нервной системы. Исключение составляет кортикоспинальные тракты (пирамидальные тракты) в мозговой ствол и иногда спинной мозг.[2]
Патология головного мозга, связанная с ЛПВП, напоминает патологию болезни Насу-Хакола (поликистозная липомембранозная остеодисплазия со склерозирующей лейкоэнцефалопатией).[7]
Диагностика
Исследования по состоянию на 2012 год включают исследования функции микроглии. Эта работа дополнительно проясняет, является ли заболевание главным образом дефектом функции микроглии. Для такого исследования микроглиальные клетки из родственных HDLS могут быть культивированы из мозга вскрытия и проанализированы в сравнении с нормальными микроглиальными клетками на основе различий в возникновении мутаций и экспрессии факторов роста.[5]
Дифференциальная диагностика
Связанные расстройства в том же спектре болезней, что и ЛПВП, включают болезнь Насу-Хакола (поликистозная липомембранозная остеодисплазия со склерозирующей лейкоэнцефалопатией ), и тип лейкодистрофия с наполненными пигментом макрофагами, называемыми пигментной ортохроматической лейкодистрофией (POLD).[3] Помимо болезни белого вещества, Насу-Хакола вызывает кисты костей. Это вызвано мутациями в генах, участвующих в одном и том же колониестимулирующий фактор (CSF) каскад сигнального пути как указано в HDLS.[8]
Болезнь Насу-Хакола, по-видимому, вызвана мутациями в белке, связывающем тирозинкиназу протеина TYRO (ТИРОБП - также известный как DAP12) или триггерный рецептор, экспрессируемый на миелоидных клетках 2 (TREM2 ) белок. В то время как различные генные мутации происходят в пути Насу-Хакола и ЛПВП, оба характеризуются дегенерацией белого вещества с аксональными сфероидами. Современные исследователи в этой области считают, что более глубокий анализ и сравнение двух генетических аномалий при этих нарушениях могут привести к лучшему пониманию механизмов заболевания при этих редких нарушениях. POLD демонстрирует невоспалительную демиелинизацию аксонов с начальными симптомами эйфории, апатии, головной боли и исполнительная дисфункция. Хотя HDLS является аутосомно-доминантным, некоторые семьи с POLD имеют особенности, которые предполагают аутосомно-рецессивное наследование.[9] Тем не менее, недавно было показано, что POLD имеет ту же генетическую основу, что и HDLS.
Клинические и генеалогические исследования
Чтобы лучше понять болезнь, исследователи ретроспективно изучили медицинские записи пробанды и другие, которые оценивались посредством клинических обследований или анкет. Образцы крови берутся из семей пробандов для генетического тестирования. Эти члены семьи оцениваются с использованием их стандартная история болезни, при прогрессировании симптомов болезни Паркинсона (Единая шкала оценки болезни Паркинсона ), а также о прогрессировании когнитивных нарушений, таких как деменция (Тест Фольштейна ).[2]
Нейровизуализация
Стандарт МРТ сканирование было выполнено на сканерах 1,5 Тл с толщиной 5 мм и интервалом 5 мм для выявления повреждений белого вещества в идентифицированных семьях. Если интенсивность сигнала МРТ-сканирования выше в областях белого вещества, чем в областях серого вещества, считается, что пациент подвержен риску HDLS, хотя ряд других заболеваний также может вызывать изменения белого вещества, и результаты не являются диагностическими без генетических тестирование или патологический подтверждение.[2]
Патология
Срезы тканей из биопсий или аутопсий головного мозга обычно встраиваются в парафин из которых вырезают срезы и устанавливают на предметные стекла для гистологических исследований. Специальные окраски на миелин и аксональную патологию показывают аномальные изменения, характерные для ЛПВП, выявляемые в белом веществе неокортекс, базальный ганглий, таламус, средний мозг, мосты и спинной мозг.[2][10] Помимо рутины гистологический методы (Окрашивание H&E ) образцы оцениваются с помощью иммуногистохимия за убиквитин, белок-предшественник амилоида и нейрофиламент для характеристики аксональных изменений и основной белок миелина для патологии миелина. Иммуногистохимические окрашивания микроглии (CD68 или HLA-DR) и астроцитов (GFAP) также являются полезными методами для характеристики патологии белого вещества.[6] Имея патологию, аналогичную POLD, HDLS обычно классифицируют как лейкоэнцефалопатию с началом у взрослых с аксональными сфероидами и пигментированной глией (ALSP), чтобы уделять этим индивидуально нераспознанным состояниям повышенное внимание.[3]
Классификация
ЛПВП попадает в категорию заболеваний белого вещества мозга, называемых лейкоэнцефалопатиями, которые характеризуются некоторой степенью дисфункции белого вещества. ЛПВП имеет поражения белого вещества с аномалиями в миелиновой оболочки вокруг аксонов, где причинные влияния постоянно исследуются на основе недавних генетических открытий. Исследования Сундала и его коллег из Швеции показали, что аллель риска у кавказцев может быть причиной, поскольку выявленные случаи до сих пор были среди больших кавказских семей.[2]
Управление
Этот раздел пуст. Вы можете помочь добавляя к этому. (Октябрь 2017 г.) |
Эпидемиология
Средний клинический профиль опубликованных исследований показывает, что средний возраст начала заболевания у пациентов с ЛПВП составляет 44,3 года при средней продолжительности заболевания 5,8 года и среднем возрасте смерти 53,2 года.[2][11] По состоянию на 2012 год было выявлено около 15 случаев, по крайней мере, из 11 спорадических случаев ЛПВП.[2][11] Случаи HDLS были обнаружены в Германии, Норвегии, Швеции и Соединенных Штатах, что свидетельствует о международном распространении, сосредоточенном между Северной Европой и США.[2]
Изучая многочисленные родственники, было установлено, что болезнь не возникала только среди мужчин или женщин, а была равномерно распределена, что свидетельствует о наличии аутосомный а не генетическое заболевание, связанное с полом. Также было замечено, что случаи ЛПВП не пропускали поколения, как это было бы при рецессивном наследовании, и поэтому были названы аутосомно-доминантными.[2]
История
Это заболевание было впервые описано в 1984 г. Аксельссоном. и другие. в большом Шведский родословная.[12] Это заболевание лучше известно невропатологам, чем клиницистам. Невропатолог, интересующийся ЛПВП, доктор Деннис В. Диксон, выявил ряд случаев невропатология исследование мозга, представленное для изучения семейной деменции и двигательных нарушений у взрослых в Нью-Йорке, а затем во Флориде. Признание важности этого расстройства как причины старческого слабоумия и двигательных нарушений было усилено в 1997 г. Клиника Майо когда доктор Збигнев К. Вшолек идентифицировал семью с ЛПВП, которая первоначально считалась следствием другого болезненного процесса (FTDP-17), но только вскрытие одного, а затем других членов семьи показали, что это ЛПВП. Wszolek основал международный консорциум в 2005 году, чтобы идентифицировать другие семьи и собрать образцы ДНК или мозга у членов семьи для нейропатологического подтверждения и генетического исследования в клинике Мэйо во Флориде.[2]
Смотрите также
Рекомендации
- ^ а б c Лин, В. Л., Вшолек, З. К., и Диксон, Д. В. (2010). Наследственная диффузная лейкоэнцефалопатия со сфероидами: ультраструктурные и иммуноэлектронные микроскопические исследования. Int J Clin Exp Pathol, 3 (7), 665-674.
- ^ а б c d е ж грамм час я j k л м Сундал, К., Лэш, Дж., Осли, Дж., Ойгарден, С., Робер, С., Кречман, Х.,. . . Wszolek, З.К. (2012). Наследственная диффузная лейкоэнцефалопатия с аксональными сфероидами (ЛПВП): заболевание, диагностированное неправильно. J Neurol Sci, 314 (1-2), 130-137. Дои:10.1016 / j.jns.2011.10.006
- ^ а б c d е ж Уидер, К., Ван Герпен, Дж. А., ДеАрмонд, С., Шустер, Э. А., Диксон, Д. В., и Вшолек, З. К. (2009). Лейкоэнцефалопатия со сфероидами (HDLS) и пигментная лейкодистрофия (POLD): единое целое? Неврология, 72 (22), 1953–1959. Дои:10.1212 / WNL.0b013e3181a826c0
- ^ а б c Радемакерс, Р., Бейкер, М., Николсон, А., Резерфорд, Н., Финч, Н., Сото-Ортолаза, А.,. . . Wszolek, Z. (2012). Мутации в рецепторе колониестимулирующего фактора 1 (CSF1R) вызывают наследственную диффузную лейкоэнцефалопатию со сфероидами. Расстройства движения, 27, S399-S400.
- ^ а б Киношита, М., Йошида, К., Оянаги, К., Хашимото, Т., и Икеда, С. (2012). Наследственная диффузная лейкоэнцефалопатия с аксональными сфероидами, вызванная мутацией R782H в CSF1R: отчет о клиническом случае. Журнал неврологических наук, 318 (1-2), 115-118. Дои:10.1016 / j.jns.2012.03.012
- ^ а б Баба Ю., Гетти Б., Бейкер М. К., Уитти Р. Дж., Хаттон М. Л., Ямагути К.,. . . Wszolek, З. К. (2006). Наследственная диффузная лейкоэнцефалопатия со сфероидами: клинические, патологические и генетические исследования нового типа. Acta Neuropathol, 111 (4), 300-311. Дои:10.1007 / s00401-006-0046-z
- ^ Хэнкок, Н., Пун, М., Тейлор, Б., и Маклин, К. (2003). Наследственная диффузная лейкоэнцефалопатия со сфероидами. J Neurol Neurosurg Psychiatry, 74 (9), 1345–1347.
- ^ Палонева, Дж., Манделин, Дж., Киялайнен, А., Бёлинг, Т., Прудло, Дж., Хакола, П.,. . . Пелтонен, Л. (2003). Дефицит DAP12 / TREM2 приводит к нарушению дифференцировки остеокластов и остеопоротических свойств. Журнал экспериментальной медицины, 198 (4), 669-675.
- ^ Knaap, Marjo S., & Valk, Jaap. (2005). Пигментная ортохроматическая лейкодистрофия Магнитный резонанс миелинизации и миелиновых расстройств (стр. 557-558): Springer Berlin Heidelberg.
- ^ Ван Герпен, Дж. А., Уидер, К., Бродерик, Д. Ф., Диксон, Д. У., Браун, Л. А., и Вшолек, З. К. (2008). Понимание динамики наследственной диффузной лейкоэнцефалопатии с аксональными сфероидами. Неврология, 71 (12), 925-929. Дои: 10.1212 / 01.wnl.0000325916.30701.21
- ^ а б Сундал, К., Ван Герпен, Дж. А., Николсон, А. М., Уидер, К., Шустер, Э. А., Ослы, Дж.,. . . Wszolek, З.К. (2012). Характеристики МРТ и оценка HDLS из-за мутаций гена CSF1R. Неврология, 79 (6), 566-574. Дои:10.1212 / WNL.0b013e318263575a
- ^ Аксельссон, Р., Ройтта, М., Сорандер, П., Акессон, Х. О., и Андерсен, О. (1984). Наследственная диффузная лейкоэнцефалопатия со сфероидами. Acta Psychiatr Scand Suppl, 314, 1-65.
внешняя ссылка
| Классификация | |
|---|---|
| Внешние ресурсы |