Формат FASTQ - FASTQ format
| Разработан | Wellcome Trust Sanger Institute |
|---|---|
| изначальный выпуск | ~2000 |
| Тип формата | Биоинформатика |
| Расширен с | ASCII и Формат FASTA |
| Интернет сайт | maq |
Формат FASTQ текстовый формат для хранения как биологической последовательности (обычно нуклеотидная последовательность ) и соответствующие показатели качества. И буква последовательности, и показатель качества кодируются одним ASCII характер для краткости.
Первоначально он был разработан в Wellcome Trust Sanger Institute связать FASTA отформатирован последовательность и ее данные о качестве, но недавно стала де-факто стандарт для хранения результатов высокопроизводительных инструментов секвенирования, таких как Иллюмина Анализатор генома.[1]
Формат
Файл FASTQ обычно использует четыре строки на последовательность.
- Строка 1 начинается с символа '@', за ней следует идентификатор последовательности и необязательный описание (как FASTA строка заголовка).
- Строка 2 - это необработанные буквы последовательности.
- Строка 3 начинается с символа "+" и необязательно за которым следует тот же идентификатор последовательности (и любое описание) снова.
- Строка 4 кодирует значения качества для последовательности в Строке 2 и должна содержать то же количество символов, что и буквы в последовательности.
Файл FASTQ, содержащий одну последовательность, может выглядеть следующим образом:
@SEQ_IDGATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT +! '' * ((((*** +)) %%% ++) (%%%%). 1 *** - + * '')) ** 55CCF >>>>>>CC65
Байт, представляющий качество, изменяется от 0x21 (самое низкое качество; '!' В ASCII) до 0x7e (самое высокое качество; '~' в ASCII). Вот символы значения качества в порядке увеличения качества слева направо (ASCII ):
! "# $% & '() * +, -. / 0123456789:; <=>? @ ABCDEFGHIJKLMNOPQRSTUVWXYZ [] ^ _` abcdefghijklmnopqrstuvwxyz {|} ~Исходные файлы Sanger FASTQ также позволяли переносить последовательность и качественные строки (разбивать на несколько строк), но это обычно не рекомендуется.[нужна цитата ] поскольку это может усложнить синтаксический анализ из-за неудачного выбора «@» и «+» в качестве маркеров (эти символы также могут встречаться в строке качества).
Идентификаторы последовательности Illumina
Последовательности из Иллюмина программное обеспечение использует систематический идентификатор:
@ HWUSI-EAS100R: 6: 73: 941: 1973 # 0/1
| HWUSI-EAS100R | уникальное название инструмента |
|---|---|
| 6 | переулок проточной ячейки |
| 73 | номер плитки в полосе проточной кюветы |
| 941 | 'x' координата кластера в тайле |
| 1973 | 'y' координата кластера в тайле |
| #0 | порядковый номер для мультиплексированной выборки (0 - без индексации) |
| /1 | член пары, / 1 или / 2 (только чтение парных или сопряженных пар) |
Версии конвейера Illumina начиная с 1.4, похоже, используют #NNNNNN вместо того #0 для идентификатора мультиплекса, где NNNNNN - это последовательность мультиплексного тега.
В Casava 1.8 формат строки '@' изменился:
@ EAS139: 136: FC706VJ: 2: 2104: 15343: 197393 1: Y: 18: ATCACG
| EAS139 | уникальное название инструмента |
|---|---|
| 136 | идентификатор запуска |
| FC706VJ | идентификатор проточной ячейки |
| 2 | переулок проточной ячейки |
| 2104 | номер плитки в полосе проточной кюветы |
| 15343 | 'x' координата кластера в тайле |
| 197393 | 'y' координата кластера в тайле |
| 1 | член пары, 1 или 2 (только чтение парных или сопряженных пар) |
| Y | Y, если чтение отфильтровано (не прошло), N иначе |
| 18 | 0, когда ни один из управляющих битов не включен, в противном случае это четное число |
| ATCACG | индексная последовательность |
Обратите внимание, что более поздние версии программного обеспечения Illumina выводят номер образца (взятый из таблицы образцов) вместо последовательности индекса. Например, в первом примере пакета может появиться следующий заголовок:
@ EAS139: 136: FC706VJ: 2: 2104: 15343: 197393 1: N: 18: 1
Архив чтения последовательности NCBI
Файлы FASTQ из INSDC Последовательность чтения из архива часто включают описание, например
@ SRR001666.1 071112_SLXA-EAS1_s_7: 5: 1: 817: длина 345 = 36GGGTGATGGCCGCTGCCGATGGCGTCAAATCCCACC + SRR001666.1 071112_SLXA-EAS1_s_7: 5: 1: 817IIIIIIIIIIIIIIIIIIII: 5: 1: 817IIIIIIIIIIIIIIIIIIII II
В этом примере есть идентификатор, присвоенный NCBI, а описание содержит исходный идентификатор из Solexa / Illumina (как описано выше) плюс длина чтения. Секвенирование выполнялось в режиме парных концов (размер вставки ~ 500 пар оснований), см. SRR001666. Формат вывода по умолчанию fastq-dump создает целые пятна, содержащие любые технические чтения и, как правило, одно- или парные биологические чтения.
$ fastq-dump.2.9.0 -Z -X 2 SRR001666Прочтите 2 места для SRR001666Написано 2 места для SRR001666@ SRR001666.1 071112_SLXA-EAS1_s_7: 5: 1: 817: 345 длина = 72GGGTGATGGCCGCTGCCGATGGCGTCAAATCCCACCAAGTTACCCTTAACAACTTAAGGGTTTTCAAATAGA+ SRR001666.1 071112_SLXA-EAS1_s_7: 5: 1: 817: 345 длина = 72IIIIIIIIIIIIIIIIIIIIIIIIIIII9IG9ICIIIIIIIIIIIIIIIIIIIIDIIIIIII> IIIIII /@ SRR001666.2 071112_SLXA-EAS1_s_7: 5: 1: 801: 338 длина = 72GTTCAGGGATACGACGTTTGTATTTTAAGAATCTGAAGCAGAAGTCGATGATAATACGCGTCGTTTTATCAT+ SRR001666.2 071112_SLXA-EAS1_s_7: 5: 1: 801: 338 длина = 72IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII6IBIIIIIIIIIIIIIIIIIIIIIIIGII> IIIII-I) 8IСовременное использование FASTQ почти всегда включает в себя разделение пятна на его биологические чтения, как описано в метаданных, предоставленных отправителем:
$ fastq-dump -X 2 SRR001666 --сплит-3Прочтите 2 места для SRR001666Написано 2 места для SRR001666$ голова SRR001666_1.fastq SRR001666_2.fastq==> SRR001666_1.fastq <==@ SRR001666.1 071112_SLXA-EAS1_s_7: 5: 1: 817: 345 длина = 36GGGTGATGGCCGCTGCCGATGGCGTCAAATCCCACC+ SRR001666.1 071112_SLXA-EAS1_s_7: 5: 1: 817: 345 длина = 36IIIIIIIIIIIIIIIIIIIIIIIIIIIIII9IG9IC@ SRR001666.2 071112_SLXA-EAS1_s_7: 5: 1: 801: 338 длина = 36GTTCAGGGATACGACGTTTGTATTTTAAGAATCTGA+ SRR001666.2 071112_SLXA-EAS1_s_7: 5: 1: 801: 338 длина = 36IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII6IBI==> SRR001666_2.fastq <==@ SRR001666.1 071112_SLXA-EAS1_s_7: 5: 1: 817: 345 длина = 36AAGTTACCCTTAACAACTTAAGGGTTTTCAAATAGA+ SRR001666.1 071112_SLXA-EAS1_s_7: 5: 1: 817: 345 длина = 36IIIIIIIIIIIIIIIIIIIIDIIIIIII> IIIIII /@ SRR001666.2 071112_SLXA-EAS1_s_7: 5: 1: 801: 338 длина = 36AGCAGAAGTCGATGATAATACGCGTCGTTTTATCAT+ SRR001666.2 071112_SLXA-EAS1_s_7: 5: 1: 801: 338 длина = 36IIIIIIIIIIIIIIIIIIIIIIGII> IIIII-I) 8IЕсли fastq-dump присутствует в архиве, он может попытаться восстановить прочитанные имена в исходный формат. NCBI по умолчанию не сохраняет исходные имена чтения:
$ fastq-dump -X 2 SRR001666 --split-3 --origfmtПрочтите 2 места для SRR001666Написано 2 места для SRR001666$ голова SRR001666_1.fastq SRR001666_2.fastq==> SRR001666_1.fastq <==@ 071112_SLXA-EAS1_s_7: 5: 1: 817: 345GGGTGATGGCCGCTGCCGATGGCGTCAAATCCCACC+ 071112_SLXA-EAS1_s_7: 5: 1: 817: 345IIIIIIIIIIIIIIIIIIIIIIIIIIIIII9IG9IC@ 071112_SLXA-EAS1_s_7: 5: 1: 801: 338GTTCAGGGATACGACGTTTGTATTTTAAGAATCTGA+ 071112_SLXA-EAS1_s_7: 5: 1: 801: 338IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII6IBI==> SRR001666_2.fastq <==@ 071112_SLXA-EAS1_s_7: 5: 1: 817: 345AAGTTACCCTTAACAACTTAAGGGTTTTCAAATAGA+ 071112_SLXA-EAS1_s_7: 5: 1: 817: 345IIIIIIIIIIIIIIIIIIIIDIIIIIII> IIIIII /@ 071112_SLXA-EAS1_s_7: 5: 1: 801: 338AGCAGAAGTCGATGATAATACGCGTCGTTTTATCAT+ 071112_SLXA-EAS1_s_7: 5: 1: 801: 338IIIIIIIIIIIIIIIIIIIIIIGII> IIIII-I) 8IВ приведенном выше примере использовались исходные имена чтения, а не присвоенное имя чтения. Запуски доступа NCBI и содержащиеся в них чтения. Исходные имена чтения, присвоенные секвенсорами, могут функционировать как локальные уникальные идентификаторы чтения и передавать столько же информации, сколько и серийный номер. Приведенные выше идентификаторы были присвоены алгоритмически на основе информации о запуске и геометрических координат. Ранние загрузчики SRA анализировали эти идентификаторы и хранили свои разложенные компоненты внутри. NCBI прекратил записывать прочитанные имена, потому что они часто изменяются по сравнению с исходным форматом поставщиков, чтобы связать некоторую дополнительную информацию, значимую для конкретного конвейера обработки, и это вызвало нарушения формата имени, что привело к большому количеству отклоненных представлений. Без четкой схемы для имен чтения их функция остается функцией уникального идентификатора чтения, передавая тот же объем информации, что и серийный номер чтения. Увидеть различные Проблемы с SRA Toolkit для подробностей и обсуждения.
Также обратите внимание, что fastq-dump преобразует эти данные FASTQ из исходной кодировки Solexa / Illumina в стандарт Sanger (см. кодировки ниже). Это потому что SRA служит хранилищем информации NGS, а не форматирует. Различные инструменты * -dump могут создавать данные в нескольких форматах из одного источника. Требования для этого были продиктованы пользователями в течение нескольких лет, причем большая часть раннего спроса исходила от Проект 1000 геномов.
Вариации
Качественный
Ценность качества Q целочисленное отображение п (т.е. вероятность того, что соответствующий базовый вызов неверен). Использовались два разных уравнения. Первый - это стандартный вариант Сэнгера для оценки надежности базового вызова, также известный как Оценка качества Phred:

Конвейер Solexa (то есть программное обеспечение, поставляемое с анализатором генома Illumina) ранее использовал другое отображение, кодирующее шансы п/(1-п) вместо вероятности п:

Хотя оба сопоставления асимптотически идентичны при более высоких значениях качества, они различаются на более низких уровнях качества (т. Е. Приблизительно п > 0,05 или эквивалентно Q < 13).
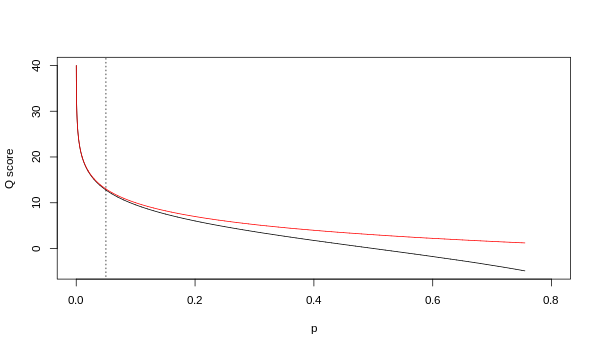
Иногда возникали разногласия относительно того, какое отображение на самом деле использует Illumina. В руководстве пользователя (Приложение B, стр. 122) для версии 1.4 конвейера Illumina указано, что: «Оценки определены как Q = 10 * log10 (p / (1-p)) [sic ], где p - вероятность вызова базы, соответствующего данной базе ".[2] Оглядываясь назад, можно сказать, что эта запись в руководстве была ошибкой. В руководстве пользователя (Что нового, стр. 5) конвейера Illumina версии 1.5 вместо этого приводится следующее описание: «Важные изменения в конвейере v1.3 [sic ]. Схема оценки качества была изменена на схему оценки Phred [то есть Sanger], кодируемую как символ ASCII путем добавления 64 к значению Phred. Оценка базы по Phred: , где е это оценочная вероятность ошибочного основания.[3]

Кодирование
- Формат Sanger может кодировать Оценка качества Phred от 0 до 93 с использованием ASCII от 33 до 126 (хотя в необработанных данных чтения оценка качества Phred редко превышает 60, более высокие оценки возможны в сборках или картах чтения). Также используется в формате SAM.[4] Согласно сообщению на форуме seqanswers.com, к концу февраля 2011 г. последняя версия (1.8) конвейера CASAVA от Illumina будет напрямую создавать fastq в формате Sanger.[5]
- Чтения PacBio HiFi, которые обычно хранятся в формате SAM / BAM, используют соглашение Сэнгера: оценки качества Phred от 0 до 93 кодируются с использованием ASCII от 33 до 126. Необработанные подпотоки PacBio используют то же соглашение, но обычно присваивают базовое качество заполнителя (Q0 ) ко всем читаемым базам.[6]
- Формат Solexa / Illumina 1.0 может кодировать показатель качества Solexa / Illumina от -5 до 62, используя ASCII От 59 до 126 (хотя в необработанных данных чтения ожидаются только оценки Solexa от -5 до 40)
- Начиная с Illumina 1.3 и до Illumina 1.8 формат закодировал Оценка качества Phred от 0 до 62 с помощью ASCII От 64 до 126 (хотя в необработанных данных чтения ожидаются только оценки Phred от 0 до 40).
- Начиная с Illumina 1.5 и до Illumina 1.8, оценки Phred от 0 до 2 имеют немного другое значение. Значения 0 и 1 больше не используются, а значение 2, закодированное ASCII 66 "B", также используется в конце чтения как Считайте индикатор контроля качества сегмента.[7] Руководство Illumina[8] (стр.30) говорится следующее: Если чтение заканчивается сегментом в основном низкого качества (Q15 или ниже), тогда все значения качества в сегменте заменяются значением 2 (закодировано как буква B в текстовой кодировке показателей качества Illumina). .. Этот индикатор Q2 не прогнозирует конкретную частоту ошибок, а скорее указывает на то, что конкретная конечная часть считывания не должна использоваться в дальнейших анализах. Кроме того, оценка качества, закодированная как буква «B», может иметь место внутри операций чтения, по крайней мере, до версии 1.6 конвейера, как показано в следующем примере:
@ HWI-EAS209_0006_FC706VJ: 5: 58: 5894: 21141 # ATCACG / 1TTAATTGGTAAATAAATCTCCTAATAGCTTAGATNTTACCTTNNNNNNNNNNTAGTTTCTTGAGATTTGTTGGGGGAGACATTTTTGTGATTGCCTTGAT + HWI-EAS209_0006_FC706VJ: 5: 58: 5894: 21141 # ATCACG / 1efcfffffcfeefffcffffffddf`feed] `] _Ba _ ^ __ [YBBBBBBBBBBRTT ]] [] dddd`ddd ^ dddadd ^ BBBBBBBBBBBBBBBBBBBBBBBBB
Была предложена альтернативная интерпретация этой кодировки ASCII.[9] Кроме того, в прогонах Illumina с использованием элементов управления PhiX наблюдалось, что символ «B» представляет «неизвестный показатель качества». Уровень ошибок при чтении «B» был примерно на 3 балла по шкале phred ниже среднего наблюдаемого балла для данного прогона.
- Начиная с Illumina 1.8, показатели качества в основном вернулись к использованию формата Sanger (Phred + 33).
Для необработанных считываний диапазон оценок будет зависеть от технологии и используемого основного вызывающего, но обычно будет до 41 для последних исследований химии Illumina. Поскольку ранее максимальная наблюдаемая оценка качества составляла всего 40, различные скрипты и инструменты ломаются, когда они сталкиваются с данными со значениями качества, превышающими 40. Для обработанных чтений оценки могут быть даже выше. Например, значения качества 45 наблюдаются при чтениях из службы последовательного считывания Illumina (ранее Moleculo).
SSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS..................................................... ..........................XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX...................... ...............................IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII...................... .................................JJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJ..................... LLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLL.................................................... PPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPP ! "# $% & '() * +, -. / 0123456789:; <=>? @ ABCDEFGHIJKLMNOPQRSTUVWXYZ [] ^ _` abcdefghijklmnopqrstuvwxyz {|} ~ | | | | | | | 33 59 64 73 104 126 0........................26...31.......40 -5....0........9.............................40 0........9.............................40 3.....9..............................41 0.2......................26...31........41 0..................20........30........40........50..........................................93
S - Sanger Phred + 33, обычно необработанные чтения (0, 40) X - Solexa Solexa + 64, обычно необработанные чтения (-5, 40) I - Illumina 1.3+ Phred + 64, обычно необработанные чтения (0, 40) J - Illumina 1.5+ Phred + 64, обычные считывания обычно (3, 41), где 0 = не используется, 1 = не используется, 2 = индикатор контроля качества сегмента чтения (жирный шрифт) (Примечание: см. Обсуждение выше). L - Illumina 1.8+ Phred + 33, обычно необработанные чтения (0, 41) P - PacBio Phred + 33, HiFi обычно читает (0, 93)
Цветовое пространство
Для данных SOLiD последовательность находится в цветовом пространстве, кроме первой позиции. Значения качества соответствуют формату Sanger. Инструменты выравнивания различаются по своей предпочтительной версии значений качества: некоторые включают оценку качества (установленную на 0, т. Е. '!') Для ведущего нуклеотида, другие - нет. Архив чтения последовательности включает этот показатель качества.
Моделирование
К моделированию чтения FASTQ подошли несколько инструментов.[10][11]Сравнение этих инструментов можно увидеть здесь.[12]
Сжатие
Общие компрессоры
Инструменты общего назначения, такие как Gzip и bzip2, рассматривают FASTQ как простой текстовый файл и приводят к неоптимальным коэффициентам сжатия. NCBI Последовательность чтения из архива кодирует метаданные с использованием схемы LZ-77. Обычные компрессоры FASTQ обычно сжимают отдельные поля (считанные имена, последовательности, комментарии и оценки качества) в файле FASTQ отдельно; к ним относятся DSRC и DSRC2, FQC, LFQC, Fqzcomp и Slimfastq.
Читает
Наличие эталонного генома удобно, потому что тогда вместо хранения самих нуклеотидных последовательностей можно просто выровнять считанные данные с эталонным геномом и сохранить позиции (указатели) и несовпадения; указатели затем могут быть отсортированы в соответствии с их порядком в эталонной последовательности и закодированы, например, с кодированием длин серий. Когда покрытие или содержание повторов в секвенированном геноме высокое, это приводит к высокой степени сжатия. СЭМ Форматы / BAM, файлы FASTQ не определяют эталонный геном. Компрессоры FASTQ на основе центровки поддерживает использование либо предоставленных пользователем, либо de novo собранная ссылка: LW-FQZip использует предоставленный эталонный геном, а Quip, Leon, k-Path и KIC выполняют de novo сборка с использованием граф де Брейна основанный на подходе.
Явное отображение чтения и de novo сборка обычно выполняется медленно. Компрессоры FASTQ на основе повторного заказа сначала кластерные чтения, которые совместно используют длинные подстроки, а затем независимо сжимают чтения в каждом кластере после их переупорядочения или сборки в более длинные контиги, достигая, возможно, наилучшего компромисса между временем работы и степенью сжатия. SCALCE - первый такой инструмент, за ним следуют Orcom и Mince. BEETL использует обобщенный Преобразование Барроуза – Уиллера для переупорядочивания чтений, а HARC обеспечивает лучшую производительность с переупорядочением на основе хэшей. AssemblTrie вместо этого собирает операции чтения в деревья ссылок с наименьшим общим числом символов в ссылке.[13][14]
Тесты для этих инструментов доступны в.[15]
Ценности качества
Значения качества составляют около половины необходимого дискового пространства в формате FASTQ (до сжатия), поэтому сжатие значений качества может значительно снизить требования к хранению и ускорить анализ и передачу данных секвенирования. В последнее время в литературе рассматриваются как сжатие без потерь, так и сжатие с потерями. Например, алгоритм QualComp [16] выполняет сжатие с потерями со скоростью (количество бит на значение качества), заданной пользователем. Основываясь на результатах теории искажения скорости, он распределяет количество битов так, чтобы минимизировать MSE (среднеквадратичную ошибку) между исходным (несжатым) и восстановленным (после сжатия) значениями качества. Другие алгоритмы сжатия значений качества включают SCALCE [17] и Fastqz.[18] Оба являются алгоритмами сжатия без потерь, которые обеспечивают дополнительный подход к управляемому преобразованию с потерями. Например, SCALCE уменьшает размер алфавита на основании наблюдения, что «соседние» значения качества в целом похожи. Для теста см..[19]
Начиная с HiSeq 2500 Illumina дает возможность выводить крупнозернистые материалы качества в бункеры качества. Разделенные баллы вычисляются непосредственно из таблицы эмпирических показателей качества, которая сама привязана к аппаратному обеспечению, программному обеспечению и химическому составу, которые использовались во время эксперимента по секвенированию.[20]
Шифрование
Шифрование файлов FASTQ в основном решается с помощью специального инструмента шифрования: Cryfa.[21] Cryfa использует шифрование AES и позволяет уплотнять данные помимо шифрования. Он также может обращаться к файлам FASTA.
Расширение файла
Нет стандарта расширение файла для файла FASTQ, но обычно используются .fq и .fastq.
Конвертеры форматов
- Биопайтон версия 1.51 и более поздние (преобразует Sanger, Solexa и Illumina 1.3+)
- EMBOSS версия 6.1.0, патч 1 и новее (преобразовывает Sanger, Solexa и Illumina 1.3+)
- BioPerl версия 1.6.1 и более поздние (конвертирует Sanger, Solexa и Illumina 1.3+)
- BioRuby версия 1.4.0 и более поздние (взаимно преобразует Sanger, Solexa и Illumina 1.3+)
- BioJava версия 1.7.1 и новее (конвертирует Sanger, Solexa и Illumina 1.3+)
Смотрите также
- В FASTA формат, используемый для представления последовательностей генома.
- В СЭМ формат, используемый для представления считываний секвенсора генома, которые были выровнены с последовательностями генома.
- В GVF формат (Genome Variation Format), расширение, основанное на GFF3 формат.
Рекомендации
- ^ Cock, P. J. A .; Филдс, С. Дж .; Goto, N .; Heuer, M. L .; Райс, П. М. (2009). «Формат файла Sanger FASTQ для последовательностей с показателями качества и варианты Solexa / Illumina FASTQ». Исследования нуклеиновых кислот. 38 (6): 1767–1771. Дои:10.1093 / нар / gkp1137. ЧВК 2847217. PMID 20015970.
- ^ Руководство пользователя программного обеспечения для анализа последовательности: для конвейерной версии 1.4 и CASAVA версии 1.0 от апреля 2009 г. PDF В архиве 10 июня 2010 г. Wayback Machine
- ^ Руководство пользователя программного обеспечения для анализа последовательности: для конвейера версии 1.5 и CASAVA версии 1.0 от августа 2009 г. PDF[мертвая ссылка ]
- ^ Формат карты последовательности / выравнивания Версия 1.0 от августа 2009 г. PDF
- ^ Тема скругляка в Seqanswer от января 2011 г. интернет сайт
- ^ Спецификация формата PacBio BAM 10.0.0 https://pacbiofileformats.readthedocs.io/en/10.0/BAM.html#qual
- ^ Показатели качества Illumina, Тобиас Манн, Bioinformatics, San Diego, Illumina http://seqanswers.com/forums/showthread.php?t=4721
- ^ Использование программного обеспечения для управления секвенированием Genome Analyzer, версия 2.6, № по каталогу SY-960-2601, № по каталогу 15009921, ред. A, ноябрь 2009 г. http://watson.nci.nih.gov/solexa/Using_SCSv2.6_15009921_A.pdf[мертвая ссылка ]
- ^ Сайт проекта SolexaQA
- ^ Хуанг, Вт; Ли, Л; Myers, J. R .; Март, Г. Т. (2012). «ART: симулятор чтения секвенирования нового поколения». Биоинформатика. 28 (4): 593–4. Дои:10.1093 / биоинформатика / btr708. ЧВК 3278762. PMID 22199392.
- ^ Пратас, Д; Пинхо, А. Дж .; Родригес, Дж. М. (2014). "XS: симулятор чтения FASTQ". BMC Research Notes. 7: 40. Дои:10.1186/1756-0500-7-40. ЧВК 3927261. PMID 24433564.
- ^ Эскалона, Мерли; Роча, Сара; Посада, Дэвид (2016). «Сравнение инструментов для моделирования геномных данных секвенирования следующего поколения». Природа Обзоры Генетика. 17 (8): 459–69. Дои:10.1038 / nrg.2016.57. ЧВК 5224698. PMID 27320129.
- ^ Гинарт А.А., Хуэй Дж., Чжу К., Нуманагич И., Куртаде Т.А., Сахиналп СК; и другие. (2018). «Оптимальное сжатое представление данных последовательности с высокой пропускной способностью с помощью легкой сборки». Nat Commun. 9 (1): 566. Bibcode:2018НатКо ... 9..566Г. Дои:10.1038 / s41467-017-02480-6. ЧВК 5805770. PMID 29422526.CS1 maint: несколько имен: список авторов (ссылка на сайт)
- ^ Чжу, Кайюань; Нуманагич, Ибрагим; Сахиналп, С. Дженк (2018). «Сжатие геномных данных». Энциклопедия технологий больших данных. Чам: Издательство Springer International. С. 779–783. Дои:10.1007/978-3-319-63962-8_55-1. ISBN 978-3-319-63962-8.
- ^ Нуманагич, Ибрагим; Бонфилд, Джеймс К; Хач, Фараз; Фогес, Ян; Остерманн, Йорн; Альберти, Клаудио; Маттавелли, Марко; Сахиналп, С. Ценк (2016-10-24). «Сравнение высокопроизводительных инструментов сжатия данных секвенирования». Природные методы. ООО "Спрингер Сайенс энд Бизнес Медиа". 13 (12): 1005–1008. Дои:10.1038 / nmeth.4037. ISSN 1548-7091. PMID 27776113. S2CID 205425373.
- ^ Очоа, Идоя; Аснани, Химаншу; Бхарадиа, Динеш; Чоудхури, Майнак; Вайсман, Цачи; Йона, Голаны (2013). "Qual Comp: Новый компрессор с потерями для оценки качества, основанный на теории искажения скорости ". BMC Bioinformatics. 14: 187. Дои:10.1186/1471-2105-14-187. ЧВК 3698011. PMID 23758828.
- ^ Hach, F; Numanagic, I; Алкан, C; Сахиналп, С. С. (2012). «SCALCE: усиление алгоритмов сжатия последовательности с использованием локально согласованного кодирования». Биоинформатика. 28 (23): 3051–7. Дои:10.1093 / биоинформатика / bts593. ЧВК 3509486. PMID 23047557.
- ^ fastqz.http://mattmahoney.net/dc/fastqz/
- ^ М. Хоссейни, Д. Пратас и А. Пиньо. 2016. Обзор методов сжатия данных для биологических последовательностей. Информация 7(4):(2016): 56
- ^ Техническая записка Illumina.http://www.illumina.com/content/dam/illumina-marketing/documents/products/technotes/technote_understanding_quality_scores.pdf
- ^ Хоссейни М., Пратас Д., Пинхо А. (2018). Cryfa: надежный инструмент шифрования геномных данных. Биоинформатика. 35. С. 146–148. Дои:10.1093 / биоинформатика / bty645. ЧВК 6298042. PMID 30020420.
внешняя ссылка
- MAQ веб-страница, обсуждающая варианты FASTQ
- Набор инструментов Fastx набор инструментов командной строки для предварительной обработки файлов Short-Reads FASTA / FASTQ
- Fastqc инструмент контроля качества для данных последовательности с высокой пропускной способностью
- GTO набор инструментов для данных FASTQ
- FastQC Fastqc на системе bwHPC-C5 в Германии
- PRINSEQ может использоваться для контроля качества и для фильтрации, переформатирования или обрезки данных последовательности (веб-версии и версии для командной строки)
- Cryfa может использоваться для безопасного шифрования файлов FASTQ, FASTA, VCF и SAM / BAM (версия для командной строки)