Гетерогенный катализ - Heterogeneous catalysis
В химии, гетерогенный катализ является катализ где фаза катализаторов отличается от катализаторов реагенты[1] или же товары. Процесс контрастирует с гомогенный катализ где реагенты, продукты и катализатор находятся в одной фазе. Фаза различает не только твердый, жидкость, и газ компоненты, но также несмешиваемый смеси (например, масло и воды ) или везде, где присутствует интерфейс. Катализаторы полезны, потому что они увеличивают скорость реакции[2] сами по себе не потребляются и поэтому могут использоваться повторно.
Гетерогенный катализ обычно включает твердофазные катализаторы и газофазные реагенты.[3] В этом случае на поверхности катализатора происходит цикл молекулярной адсорбции, реакции и десорбции. Термодинамика, массообмен и теплопередача влияют на скорость (кинетика) реакции.
Гетерогенный катализ очень важен, поскольку он обеспечивает более быстрое крупномасштабное производство и селективное образование продукта.[4] Примерно 35% мирового ВВП находится под влиянием катализа.[5] Производство 90% химикатов (по объему) обеспечивается твердыми катализаторами.[3] Химическая и энергетическая промышленность в значительной степени полагаются на гетерогенный катализ. Например, Процесс Габера-Боша использует катализаторы на основе металлов в синтезе аммиак, важный компонент удобрений; В 2016 году произведено 144 млн тонн аммиака.[6]
Адсорбция
Адсорбция является важным этапом гетерогенного катализа. Адсорбция - это процесс, при котором молекула в газовой (или растворной) фазе (адсорбат) связывается с твердыми (или жидкими) поверхностными атомами (адсорбентом). Обратной адсорбции является десорбция, отделение адсорбата от адсорбента. В реакции, которой способствует гетерогенный катализ, катализатор является адсорбентом, а реагенты - адсорбатом.
Типы адсорбции
Различают два типа адсорбции: физическая адсорбция, слабосвязанная адсорбция и хемосорбция, прочно связанная адсорбция. Многие процессы гетерогенного катализа находятся между двумя крайностями. В Модель Леннарда-Джонса обеспечивает базовую основу для предсказания молекулярных взаимодействий как функции разделения атомов.[7]
Физисорбция
При физадсорбции молекула притягивается к поверхностным атомам через силы Ван дер Ваальса. К ним относятся диполь-дипольные взаимодействия, индуцированные дипольные взаимодействия и дисперсионные силы Лондона. Обратите внимание, что между адсорбатом и адсорбентом не образуются химические связи, и их электронные состояния остаются относительно неизменными. Типичные энергии для физической адсорбции составляют от 3 до 10 ккал / моль.[3] При гетерогенном катализе, когда молекула реагента физадсорбируется на катализаторе, обычно говорят, что она находится в состоянии предшественника, промежуточном энергетическом состоянии перед хемосорбцией, при более прочно связанной адсорбции.[7] Из состояния-предшественника молекула может подвергаться хемосорбции, десорбции или миграции по поверхности.[8] Природа состояния предшественника может влиять на кинетику реакции.[8]
Хемосорбция
Когда молекула приближается к поверхностным атомам настолько близко, что их электронные облака перекрытие может произойти хемосорбция. При хемосорбции адсорбат и адсорбент разделяют электроны, что означает образование химические связи. Типичные энергии хемосорбции находятся в диапазоне от 20 до 100 ккал / моль.[3] Два случая хемосорбции:
- Молекулярная адсорбция: адсорбат остается неповрежденным. Примером является связывание алкена платиной.
- Диссоциативная адсорбция: одна или несколько связей разрываются одновременно с адсорбцией. В этом случае барьер для диссоциация влияет на скорость адсорбции. Примером этого является связывание H2 к металлическому катализатору, где связь H-H разрывается при адсорбции.
Поверхностные реакции

Большинство реакций на поверхности металлов происходят распространение цепи в котором каталитические промежуточные продукты производятся и потребляются циклически.[9] Можно описать два основных механизма поверхностных реакций для A + B → C.[3]
- Механизм Ленгмюра-Хиншелвуда: молекулы реагента А и В адсорбируются на каталитической поверхности. Адсорбируясь на поверхности, они объединяются с образованием продукта C, который затем десорбируется.
- Механизм Элея-Ридила: одна молекула реагента, А, адсорбируется на каталитической поверхности. Без адсорбции B реагирует с абсорбированным A с образованием C, который затем десорбируется с поверхности.
Наиболее гетерогенно катализируемые реакции описываются моделью Ленгмюра-Хиншелвуда.[10]
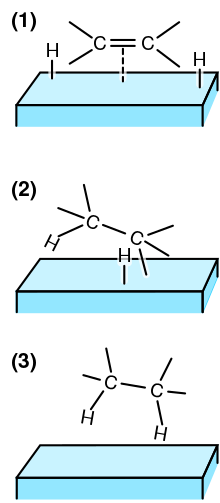
При гетерогенном катализе реагенты размытый из основной жидкой фазы в адсорбировать к поверхности катализатора. Сайт адсорбции не всегда является активным сайтом катализатора, поэтому молекулы реагента должны мигрировать по поверхности к активному центру. В активном центре молекулы реагента будут реагировать с образованием молекулы (-ов) продукта, следуя более энергетически легкому пути через каталитические промежуточные соединения (см. Рисунок справа). Затем молекулы продукта десорбируются с поверхности и диффундируют. Сам катализатор остается нетронутым и свободен для проведения дальнейших реакций. Явления переноса, такие как тепломассоперенос, также играют роль в наблюдаемой скорости реакции.
Дизайн катализатора
Катализаторы не активны по отношению к реагентам по всей своей поверхности; только определенные места обладают каталитической активностью, называемой активные сайты. Площадь поверхности твердого катализатора сильно влияет на количество доступных активных центров. В промышленной практике твердые катализаторы часто бывают пористыми для увеличения площади поверхности, обычно достигающей 50–400 мкм.2/грамм.[3] Немного мезопористые силикаты, такие как MCM-41, имеют площадь поверхности более 1000 м2/грамм.[11] Пористые материалы экономически эффективны из-за их высокого отношения площади поверхности к массе и повышенной каталитической активности.
Во многих случаях твердый катализатор рассредоточенный на поддерживающем материале для увеличения площади поверхности (увеличения числа активных центров) и обеспечения стабильности.[3] Обычно носители для катализаторов являются инертными материалами с высокой температурой плавления, но сами могут быть каталитическими. Большинство носителей катализаторов являются пористыми (часто на основе углерода, диоксида кремния, цеолита или оксида алюминия).[5] и выбран из-за их высокого отношения площади поверхности к массе. Для данной реакции необходимо выбирать пористые носители, чтобы реагенты и продукты могли входить в материал и выходить из него.
Часто вещества намеренно добавляются к реакционному сырью или к катализатору, чтобы повлиять на каталитическую активность, селективность и / или стабильность. Эти соединения называются промоторами. Например, оксид алюминия (Al2О3) добавляется во время синтеза аммиака для обеспечения большей стабильности за счет замедления процессов спекания на Fe-катализаторе.[3]
Принцип Сабатье можно рассматривать как один из краеугольных камней современной теории катализа.[12] Принцип Сабатье гласит, что взаимодействие поверхности с адсорбатами должно быть оптимальным: не слишком слабым, чтобы быть инертным по отношению к реагентам, и не слишком сильным, чтобы отравить поверхность и избежать десорбции продуктов.[13] Утверждение о том, что взаимодействие поверхности с адсорбатом должно быть оптимальным, носит качественный характер. Обычно количество адсорбатов и переходных состояний, связанных с химической реакцией, велико, поэтому оптимальный нужно найти в многомерном пространстве. Разработка катализатора в таком многомерном пространстве не является вычислительно жизнеспособной задачей. Кроме того, такой процесс оптимизации будет далеко не интуитивно понятным. Соотношения масштабирования используются для уменьшения размерности пространства конструкции катализатора.[14] Такие соотношения являются корреляциями между энергиями связи адсорбатов (или между энергиями связи адсорбата и переходными состояниями, также известными как BEP отношения )[15] которые «достаточно похожи», например, масштабирование OH и OOH.[16] Применение масштабных соотношений к задачам конструкции катализатора значительно снижает размерность пространства (иногда до 1 или 2).[17] Можно также использовать микрокинетическое моделирование, основанное на таких масштабных соотношениях, чтобы учесть кинетику, связанную с адсорбцией, реакцией и десорбцией молекул при определенных условиях давления или температуры.[18] Такое моделирование затем приводит к хорошо известным вулканическим сюжетам, на которых оптимум, качественно описанный принципом Сабатье, упоминается как «вершина вулкана». Соотношения масштабирования можно использовать не только для подключения энергетики радикальный поверхностно-адсорбированные группы (например, O *, OH *),[14] но также для подключения энергетики закрытая оболочка молекулы между собой или с соответствующими адсорбатами радикалов.[19] В последнее время перед исследователями каталитических наук стоит задача «разорвать» отношения масштабирования.[20] Корреляции, которые проявляются в соотношениях масштабирования, ограничивают пространство конструкции катализатора, не позволяя достичь «вершины вулкана». Нарушение отношений масштабирования может относиться либо к проектированию поверхностей или мотивов, которые не следуют соотношению масштабирования, либо к тем, которые следуют другому соотношению масштабирования (чем обычное соотношение для связанных адсорбатов) в правильном направлении: такое, которое может приблизить нас к вершина вулкана реактивности.[17] Помимо изучения каталитической реакционной способности, масштабные соотношения могут использоваться для изучения и отбора материалов на предмет селективности по отношению к конкретному продукту.[21] Существуют особые комбинации энергий связывания, которые отдают предпочтение определенным продуктам по сравнению с другими. Иногда набор энергий связывания, которые могут изменить селективность по отношению к конкретному продукту, «масштабируется» друг с другом, таким образом, чтобы улучшить селективность, необходимо нарушить некоторые соотношения масштабирования; Примером этого является масштабирование между энергиями окислительной активации метана и метанола, что приводит к отсутствию селективности при прямом преобразовании метана в метанол.[22]
Деактивация катализатора
Дезактивация катализатора определяется как потеря каталитической активности и / или селективности с течением времени.
Вещества, снижающие скорость реакции, называются яды. Яды хемосорбируются на поверхности катализатора и уменьшают количество доступных активных центров для связывания молекул реагента.[23] Обычные яды включают элементы групп V, VI и VII (например, S, O, P, Cl), некоторые токсичные металлы (например, As, Pb) и адсорбирующие вещества с множественными связями (например, CO, ненасыщенные углеводороды).[7][23] Например, сера нарушает производство метанола, отравляя катализатор Cu / ZnO.[24] Вещества, повышающие скорость реакции, называются промоутеры. Например, присутствие щелочных металлов в синтезе аммиака увеличивает скорость N2 диссоциация.[24]
Присутствие ядов и промоторов может изменить энергию активации стадии, ограничивающей скорость, и повлиять на селективность катализатора в отношении образования определенных продуктов. В зависимости от количества вещество может быть благоприятным или неблагоприятным для химического процесса. Например, при производстве этилена небольшое количество хемосорбированного хлора будет действовать как промотор, улучшая селективность Ag-катализатора в отношении этилена по сравнению с CO.2, а слишком много хлора будет действовать как яд.[7]
Другие механизмы дезактивации катализатора включают:
- Спекание: при нагревании диспергированные частицы каталитического металла могут перемещаться по поверхности носителя и образовывать кристаллы. Это приводит к уменьшению площади поверхности катализатора.
- Обрастание: осаждение материалов из жидкой фазы на твердофазный катализатор и / или поверхности носителя. Это приводит к закупорке активного сайта и / или пор.
- Коксование: отложение тяжелых, богатых углеродом твердых частиц на поверхности из-за разложения углеводородов[23]
- Реакции пар-твердое тело: образование неактивного поверхностного слоя и / или образование летучего соединения, которое выходит из реактора.[23] Это приводит к потере площади поверхности и / или материала катализатора.
- Твердотельное преобразование: твердофазная диффузия атомов носителя катализатора к поверхности с последующей реакцией, которая образует неактивную фазу. Это приводит к потере площади поверхности катализатора.
- Эрозия: постоянное истирание материала катализатора, обычное в реакторах с псевдоожиженным слоем.[25] Это приводит к потере материала катализатора.
В промышленности дезактивация катализатора обходится в миллиарды долларов каждый год из-за остановки процесса и замены катализатора.[23]
Промышленные примеры
В промышленности необходимо учитывать многие конструктивные параметры, включая конструкцию реактора и катализатора в различных масштабах, от субнанометра до десятков метров. Обычные реакторы гетерогенного катализа включают: партия, непрерывный, и реакторы с псевдоожиженным слоем, в то время как более поздние установки включают стационарный, микроканальный и многофункциональный реакторы.[7] Другими переменными, которые следует учитывать, являются размеры реактора, площадь поверхности, тип катализатора, носитель катализатора, а также рабочие условия реактора, такие как температура, давление и концентрации реагентов.

Некоторые крупномасштабные промышленные процессы с использованием гетерогенных катализаторов перечислены ниже.[5]
| Процесс | Реагенты, продукт / ы (несбалансированные) | Катализатор | Комментарий |
|---|---|---|---|
| Синтез серной кислоты (Контактный процесс ) | ТАК2 + O2, ТАК3 | оксиды ванадия | Гидратация SO3 дает H2ТАК4 |
| Синтез аммиака (Процесс Габера – Боша ) | N2 + H2, NH3 | оксиды железа на глинозем (Al2О3) | Потребляет 1% мирового бюджета промышленной энергии[3] |
| Синтез азотной кислоты (Оствальдский процесс ) | NH3 + O2, HNO3 | марля Pt-Rh без подложки | Прямые маршруты из N2 неэкономичны |
| Производство водорода Паровой риформинг | CH4 + H2ОЙ2 + CO2 | Никель или К2О | Более экологичные маршруты до H2 к расщепление воды активно искал |
| Окись этилена синтез | C2ЧАС4 + O2, С2ЧАС4О | серебро на глинозем, со многими промоутерами | Плохо применимо к другим алкенам |
| Синтез цианистого водорода (Андрусовское окисление ) | NH3 + O2 + CH4, HCN | Pt-Rh | Связанный аммоксидирование процесс превращает углеводороды в нитрилы |
| Полимеризация олефинов Полимеризация Циглера-Натта | пропилен, полипропилен | TiCl3 на MgCl2 | Существует множество вариантов, в том числе некоторые однородные примеры |
| Обессеривание нефти (гидрообессеривание ) | ЧАС2 + R2S (идеализированная сероорганическая примесь), RH + H2S | Пн -Co на глиноземе | Добывает углеводороды с низким содержанием серы, сера извлекается из Процесс Клауса |

Другие примеры
- Сокращение нитрилы в синтезе фенэтиламин с Никель Ренея катализатор и водород в аммиак:[26]
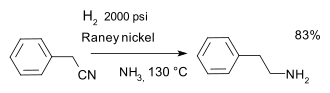 Гидрирование нитрила
Гидрирование нитрила - Растрескивание, изомеризация, и реформирование углеводороды для образования подходящих и полезных смесей бензина.
- В автомобилях, каталитические преобразователи используются для катализирования трех основных реакций:
- В окисление из монооксид углерода к углекислый газ:
- 2CO (г) + O2(г) → 2CO2(грамм)
- В снижение из окись азота вернуться к азот:
- 2НО (г) + 2СО (г) → N2(г) + 2CO2(грамм)
- В окисление из углеводороды поливать и углекислый газ:
- 2 С6ЧАС6 + 15 O2 → 12 СО2 + 6 часов2О
- В окисление из монооксид углерода к углекислый газ:
- Этот процесс может происходить с любым из углеводород, но чаще всего выполняется с бензин или же дизель.
- Асимметричный гетерогенный катализ облегчает получение чистых энантиомерных соединений с использованием хиральных гетерогенных катализаторов.[27]
- Подавляющее большинство гетерогенных катализаторов основано на металлы[28] или же оксиды металлов;[29][30] однако некоторые химические реакции могут быть катализируется углеродом материалы на основе, например, окислительные дегидрирование[31] или выборочный окисления.[32]
- Этилбензол + 1/2 O2 → Стирол + H2О
- Акролеин + 1/2 O2 → Акриловая кислота

Катализируемые реакции твердое тело-жидкость и жидкость-жидкость
Хотя большинство гетерогенных катализаторов представляют собой твердые частицы, существует несколько вариантов, которые имеют практическую ценность. В случае двух несмешивающихся растворов (жидкостей) один несет катализатор, а другой - реагент. Эта установка является основой двухфазного катализа, применяемого в промышленном производстве бутиральдегида путем гидроформилирования пропилена.[33]
| Фазы реакции | Приведены примеры | Комментарий |
|---|---|---|
| твердый + раствор | гидрирование жирных кислот никелем | используется для производства маргарин |
| несмешивающиеся жидкие фазы | гидроформилирование из пропен | катализатор водной фазы; реагенты и продукты в основном в неводной фазе |
Смотрите также
- Гетерогенный золотой катализ
- Катализаторы на основе наноматериалов
- Наночастицы платины
- Снижение с программированием температуры
- Термодесорбционная спектроскопия
Рекомендации
- ^ Шлёгль, Роберт (2015-03-09). «Гетерогенный катализ». Angewandte Chemie International Edition. 54 (11): 3465–3520. Дои:10.1002 / anie.201410738. HDL:11858 / 00-001M-0000-0025-0A33-6. PMID 25693734.
- ^ Химия, Международный союз теоретических и прикладных наук. «Золотая книга ИЮПАК - катализатор». goldbook.iupac.org. Получено 2019-02-12.
- ^ а б c d е ж грамм час я Ротенберг, Гади (2008). Катализ: концепции и экологические приложения. Вайнхайм [Германия]: Wiley-VCH. ISBN 9783527318247. OCLC 213106542.
- ^ Информация., Национальная лаборатория Лоуренса Беркли. Соединенные Штаты. Министерство энергетики. Управление научно-технической информации (2003 г.). Влияние нанонауки на гетерогенный катализ. Национальная лаборатория Лоуренса Беркли. OCLC 727328504.
- ^ а б c Ма, Чжэнь; Заера, Франциско (2006-03-15), «Гетерогенный катализ металлами», в Кинге, Р. Брюсе; Крэбтри, Роберт Х .; Lukehart, Charles M .; Этвуд, Дэвид А. (ред.), Энциклопедия неорганической химии, John Wiley & Sons, Ltd, Дои:10.1002 / 0470862106.ia084, ISBN 9780470860786
- ^ "Геологическая служба США, сводки по минеральным сырьевым товарам" (PDF). USGS. Январь 2018.
- ^ а б c d е Thomas, J.M .; Томас, В. Дж. (19 ноября 2014 г.). Принципы и практика гетерогенного катализа (Вторая, переработанная ред.). Вайнхайм, Германия. ISBN 9783527683789. OCLC 898421752.
- ^ а б Боукер, Майкл (2016-03-28). «Роль состояний-предшественников в адсорбции, поверхностных реакциях и катализе». Темы в катализе. 59 (8–9): 663–670. Дои:10.1007 / s11244-016-0538-6. ISSN 1022-5528. PMID 21386456.
- ^ Р. И. Масел, «Принципы адсорбции и реакции на твердых поверхностях», Серия Wiley в области химической инженерии, Wiley-Interscience, Нью-Йорк, США, 1996, ISBN 978-0-471-30392-3
- ^ Петухов, А. (1997). «Влияние молекулярной подвижности на кинетику электрохимической реакции Ленгмюра-Хиншелвуда». Письма по химической физике. 277 (5–6): 539–544. Дои:10.1016 / с0009-2614 (97) 00916-0. ISSN 0009-2614.
- ^ Kresge, C.T .; Леонович, М. Э .; Roth, W. J .; Vartuli, J.C .; Бек, Дж. С. (1992). «Заказанные мезопористые молекулярные сита, синтезированные по механизму жидкокристаллического темплата». Природа. 359 (6397): 710–712. Bibcode:1992Натура.359..710K. Дои:10.1038 / 359710a0. ISSN 0028-0836. S2CID 4249872.
- ^ Медфорд, Эндрю Дж .; Воеводич, Александра; Hummelshøj, Jens S .; Восс, Йоханнес; Абильд-Педерсен, Франк; Студт, Феликс; Блигаард, Томас; Нильссон, Андерс; Нёрсков, Йенс К. (2015). «От принципа Сабатье к предсказательной теории гетерогенного катализа переходных металлов». Журнал катализа. 328: 36–42. Дои:10.1016 / j.jcat.2014.12.033.
- ^ Лаурсен, Андерс Б .; Мужчина, Изабела Костинела; Trinhammer, Ole L .; Россмейсл, Ян; Даль, Сорен (2011-10-04). «Принцип Сабатье, иллюстрируемый каталитическим разложением H2O2 на металлических поверхностях». Журнал химического образования. 88 (12): 1711–1715. Дои:10.1021 / ed101010x.
- ^ а б Abild-Pedersen, F .; Greeley, J .; Studt, F .; Rossmeisl, J .; Munter, T. R .; Моисей, П.Г .; Skúlason, E .; Bligaard, T .; Нёрсков, Дж. К. (2007-07-06). «Масштабные свойства энергий адсорбции водородсодержащих молекул на поверхностях переходных металлов» (PDF). Письма с физическими проверками. 99 (1): 016105. Дои:10.1103 / PhysRevLett.99.016105. PMID 17678168.
- ^ Nørskov, Jens K .; Christensen, Claus H .; Блигаард, Томас; Мюнтер, Туре Р. (18 августа 2008 г.). «Соотношения BEP для диссоциации N2 на ступенчатых поверхностях переходных металлов и сплавов». Физическая химия Химическая физика. 10 (34): 5202–5206. Дои:10.1039 / B720021H. ISSN 1463-9084. PMID 18728861.
- ^ Вишванатан, Венкатасубраманиан; Хансен, Гейне Антон; Россмейсл, Ян; Нёрсков, Йенс К. (11.07.2012). «Универсальность электрокатализа восстановления кислорода на металлических поверхностях». Катализ ACS. 2 (8): 1654–1660. Дои:10.1021 / cs300227s. ISSN 2155-5435.
- ^ а б Nørskov, Jens K .; Воеводич, Александра (01.06.2015). «Новая парадигма проектирования гетерогенных катализаторов». Национальный научный обзор. 2 (2): 140–143. Дои:10.1093 / nsr / nwv023. ISSN 2095-5138.
- ^ Медфорд, Эндрю Дж .; Ши, Чуань; Хоффманн, Макс Дж .; Lausche, Adam C .; Фитцгиббон, Шон Р.; Блигаард, Томас; Нёрсков, Йенс К. (01.03.2015). «CatMAP: программный пакет для микрокинетического картирования на основе дескрипторов каталитических тенденций». Письма о катализе. 145 (3): 794–807. Дои:10.1007 / s10562-015-1495-6. ISSN 1572-879X. S2CID 98391105.
- ^ Какехани, Арвин; Ролинг, Люк Т .; Кулькарни, Амбариш; Latimer, Allegra A .; Аброшан, Хади; Шуман, Юлия; Аль-Джама, Хасан; Сиахростами, Самира; Исмаил-Бейги, Сохраб (18.06.2018). "Природа связей неподеленная пара – поверхность и их масштабные отношения". Неорганическая химия. 57 (12): 7222–7238. Дои:10.1021 / acs.inorgchem.8b00902. ISSN 0020-1669. OSTI 1459598. PMID 29863849.
- ^ Чен, Пинг; Он, Тэн; Ву, Гуотао; Го, Цзяньпин; Гао, Вэньбо; Чанг, Фэй; Ван, Пэйкунь (январь 2017 г.). «Нарушение масштабных соотношений для достижения низкотемпературного синтеза аммиака посредством LiH-опосредованного переноса азота и гидрирования». Химия природы. 9 (1): 64–70. Дои:10.1038 / nchem.2595. ISSN 1755-4349. PMID 27995914.
- ^ Шуман, Юлия; Медфорд, Эндрю Дж .; Ю, Чон Сок; Чжао, Чжи-Цзянь; Ботра, Паллави; Цао, Анг; Студт, Феликс; Абильд-Педерсен, Франк; Нёрсков, Йенс К. (13 марта 2018 г.). «Селективность превращения синтез-газа в оксигенаты C2 + на поверхностях переходных металлов с ГЦК (111)». Катализ ACS. 8 (4): 3447–3453. Дои:10.1021 / acscatal.8b00201. OSTI 1457170.
- ^ Nørskov, Jens K .; Студт, Феликс; Абильд-Педерсен, Франк; Цай, Чарли; Ю, Чон Сок; Монтойя, Джозеф Х .; Алджама, Хасан; Кулкарни, Амбариш Р .; Латимер, Аллегра А. (февраль 2017 г.). «Понимание тенденций активации связи C – H в гетерогенном катализе». Материалы Природы. 16 (2): 225–229. Дои:10.1038 / nmat4760. ISSN 1476-4660. PMID 27723737.
- ^ а б c d е Варфоломей, Кальвин Х (2001). «Механизмы дезактивации катализатора». Прикладной катализ A: Общие. 212 (1–2): 17–60. Дои:10.1016 / S0926-860X (00) 00843-7.
- ^ а б Нёрсков, Йенс К. (25 августа 2014 г.). Основные концепции гетерогенного катализа. Studt, Felix., Abild-Pedersen, Frank., Bligaard, Thomas. Хобокен, Нью-Джерси. ISBN 9781118892022. OCLC 884500509.
- ^ Форзатти, П (1999-09-14). «Деактивация катализатора». Катализ сегодня. 52 (2–3): 165–181. Дои:10.1016 / s0920-5861 (99) 00074-7. ISSN 0920-5861.
- ^ Organic Syntheses, Coll. Vol. 3, стр.720 (1955); Vol. 23, стр.71 (1943). https://web.archive.org/web/20120315000000*/http://orgsynth.org/orgsyn/pdfs/CV4P0603.pdf
- ^ Хейтбаум; Глориус; Эшер (2006). «Асимметричный гетерогенный катализ». Энгью. Chem. Int. Эд. 45 (29): 4732–62. Дои:10.1002 / anie.200504212. PMID 16802397.
- ^ Ван, Айцинь; Ли, Цзюнь; Чжан, Тао (июнь 2018 г.). «Гетерогенный одноатомный катализ». Обзоры природы Химия. 2 (6): 65–81. Дои:10.1038 / s41570-018-0010-1. ISSN 2397-3358. S2CID 139163163.
- ^ Цзэн, Лян; Чэн, Чжо; Fan, Jonathan A .; Фань, Лян-Ши; Гонг, Цзиньлун (ноябрь 2018 г.). «Редокс-химия оксидов металлов для химических циклических процессов». Обзоры природы Химия. 2 (11): 349–364. Дои:10.1038 / s41570-018-0046-2. ISSN 2397-3358. S2CID 85504970.
- ^ Науманн д'Алнонкур, Рауль; Чепеи, Ленард-Иштван; Хэвекер, Майкл; Girgsdies, Франк; Schuster, Manfred E .; Шлёгль, Роберт; Траншке, Аннетт (2014). «Реакционная сеть при окислении пропана на фазово-чистых оксидных катализаторах MoVTeNb M1». Дж. Катал. 311: 369–385. Дои:10.1016 / j.jcat.2013.12.008. HDL:11858 / 00-001M-0000-0014-F434-5.
- ^ Zhang, J .; Лю, X .; Blume, R .; Чжан, А .; Schlögl, R .; Су, Д. С. (2008). «Поверхностно-модифицированные углеродные нанотрубки катализируют окислительное дегидрирование н-бутана». Наука. 322 (5898): 73–77. Bibcode:2008Научный ... 322 ... 73Z. Дои:10.1126 / science.1161916. HDL:11858 / 00-001M-0000-0010-FE91-E. PMID 18832641. S2CID 35141240.
- ^ Франк, B .; Blume, R .; Ринальди, А .; Trunschke, A .; Шлёгль Р. (2011). "Кислородный катализатор внедрения", sp.2 Углерод ». Энгью. Chem. Int. Эд. 50 (43): 10226–10230. Дои:10.1002 / anie.201103340. PMID 22021211.
- ^ Бой Корнилс; Вольфганг А. Херрманн, ред. (2004). Металлоорганический катализ в водной фазе: концепции и применение. Wiley-VCH.
внешняя ссылка
 СМИ, связанные с Гетерогенный катализ в Wikimedia Commons
СМИ, связанные с Гетерогенный катализ в Wikimedia Commons
| Авторитетный контроль |
|---|