Реакция Джонсона – Кори – Чайковского - Johnson–Corey–Chaykovsky reaction
| Реакция Джонсона-Кори-Чайковского | |
|---|---|
| Названный в честь | А. Уильям Джонсон Элиас Джеймс Кори Михаил Чайковский |
| Тип реакции | Реакция образования кольца |
| Идентификаторы | |
| Портал органической химии | Кори-Чайковский-реакция |
В Реакция Джонсона – Кори – Чайковского (иногда называемый Реакция Кори – Чайковского или CCR) это химическая реакция используется в органическая химия для синтеза эпоксиды, азиридины, и циклопропаны. Он был открыт в 1961 году А. Уильямом Джонсоном и значительно развит Э. Дж. Кори и Михаил Чайковский. Реакция включает добавление серы. илида к кетон, альдегид, я добываю, или Enone с образованием соответствующего 3-членного кольца. Реакция диастереоселективный благоприятствуя транс замена в товаре независимо от начального стереохимия. Синтез эпоксиды этот метод служит важным ретросинтетический альтернатива традиционному эпоксидирование реакции олефины.

Реакция чаще всего используется для эпоксидирования через метилен передачи, и с этой целью был использован в нескольких известных общий синтез (Увидеть Синтез эпоксидов ниже). Кроме того, ниже подробно описаны история, механизм, объем и энантиоселективные варианты реакции. Опубликовано несколько обзоров.[1][2][3][4][5]
История
Первоначальная публикация Джонсона касалась реакции флуоренилида 9-диметилсульфония с замещенным бензальдегид производные. Попытка Реакция типа Виттига не удалось, и вместо этого был получен оксид бензалфлуорена, при этом было отмечено, что «реакция между илидом серы и бензальдегидами не дает бензальфлуоренов, в отличие от илидов фосфора и мышьяка».[6]

Последующее развитие (диметилоксосульфанил) метанида, (CH3)2СОЧ2 и (диметилсульфанил) метанид, (CH3)2SCH2 (известный как Кори – Чайковский реактивы) Кори и Чайковский в качестве эффективных реагентов для переноса метилена установили, что реакция является частью органического канона.[7]

Механизм
В механизм реакции для реакции Джонсона – Кори – Чайковского состоит из нуклеофильное присоединение из илида к карбонил или я добываю группа. Отрицательный заряд переносится на гетероатом и потому что сульфоний катион хороший уходящая группа он выталкивается, образуя кольцо. В связанных Реакция Виттига, образование гораздо более сильных фосфор -кислород двойная связь предотвращает оксиран формирование и вместо этого олефинирование происходит через 4-членный циклический интермедиат.[4]

В транс диастереоселективность наблюдаемые результаты из-за обратимости начального добавления, что позволяет уравновешивать предпочтительный анти бетаин над син бетаин. Первоначальное добавление илида приводит к бетаину с соседними зарядами; теория функционала плотности расчеты показали, что ограничивающий шаг поворот центральной связи в конформер, необходимый для атака сзади по сульфонию.[1]

Степень обратимости на начальном этапе (и, следовательно, диастереоселективность) зависит от четырех факторов, причем большая обратимость соответствует более высокой селективности:[1]
- Устойчивость основания с более высокой стабильностью, ведущей к большей обратимости за счет предпочтения исходного материала по сравнению с бетаином.
- Стабильность илида с более высокой стабильностью, что также ведет к большей обратимости.
- Стерическое препятствие в бетаине с большим препятствием, ведущим к большей обратимости, поскольку препятствует образованию промежуточного соединения и замедляет лимитирующее скорость вращения центральной связи.
- Решение сборов в бетаине от противоионы такие как литий с большей сольватацией, позволяющей более легкое вращение в промежуточном бетаине, снижая степень обратимости.
Объем
Применение реакции Джонсона – Кори – Чайковского в органическом синтезе разнообразно. Реакция стала охватывать реакции многих типов илидов серы с электрофилы далеко за пределами оригинальных публикаций. Он нашел применение в ряде громких тотальных синтезов, как подробно описано ниже, и, как правило, признан мощным трансформирующим инструментом в органическом репертуаре.
Виды илидов

Многие типы илидов могут быть получены с различными функциональными группами как на анионном углеродном центре, так и на сере. Схема замещения может влиять на легкость приготовления реагентов (обычно из галогенида сульфония, например иодид триметилсульфония ) и общую скорость реакции различными способами. Общий формат реагента показан справа.[1]
Использование сульфоксония позволяет более легко приготовить реагент с использованием более слабых оснований по сравнению с илидами сульфония. (Разница в том, что сульфоксоний содержит кислород с двойной связью, а сульфоний - нет.) Первые реагируют медленнее из-за их повышенной стабильности. В дополнение диалкилсульфоксид побочные продукты реагентов сульфоксония намного предпочтительнее значительно более токсичных, летучих и пахучих диалкилсульфид побочные продукты сульфониевых реагентов.[1]
Подавляющее большинство реагентов монозамещены по илиду углерода (либо R1 или R2 как водород). Дизамещенные реагенты встречаются гораздо реже, но описаны:[1]
- Если илидный углерод замещен электроноакцепторная группа (EWG) реагент обозначается как стабилизированный илид. Они, как и реагенты сульфоксония, реагируют намного медленнее и, как правило, их легче приготовить. Их полезность ограничена, поскольку реакция может стать чрезмерно вялой: примеры, включающие амиды широко распространены, гораздо реже вовлекают сложные эфиры и практически нет примеров с участием других EWG. Для них соответствующие Реакция Дарценса обычно более уместен.
- Если илидный углерод замещен арил или аллил группы реагент обозначается как полустабилизированный илид. Они получили широкое развитие, уступая только классическим метилен реагенты (R1= R2= H). Характер замещения арильных реагентов может сильно влиять на селективность реакции в соответствии с вышеуказанными критериями.
- Если илид углерода замещен алкильной группой, реагент упоминается как нестабилизированный илид. Размер алкильных групп является основным фактором селективности по отношению к этим реагентам.
R-группы на сере, хотя обычно метилы, были использованы для синтеза реагентов, которые могут выполнять энантиоселективный варианты реакции (см. Варианты ниже). Размер групп также может влиять на диастереоселективность в алициклический субстраты.[1]
Синтез эпоксидов
Реакции сернистых илидов с кетоны и альдегиды формировать эпоксиды на сегодняшний день являются наиболее частым применением реакции Джонсона – Кори – Чайковского. Сообщалось о примерах, включающих сложные субстраты и «экзотические» илиды, как показано ниже.[8][9]

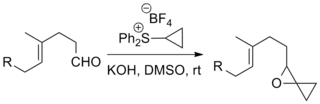
Реакция была использована в ряде известных полных синтезов, включая Полный синтез таксола Данишефского, что дает химиотерапевтический препарат, средство, медикамент таксол, а Кюне Стрихнин полный синтез который производит пестицид стрихнин.[10][11]


Синтез азиридинов
Синтез азиридины от имины является еще одним важным приложением реакции Джонсона – Кори – Чайковского и предлагает альтернативу амин перевод из оксазиридины. Хотя эта реакция менее широко применяется, она имеет аналогичный объем субстрата и функциональная группа толерантность к карбонильному эквиваленту. Примеры, показанные ниже, являются репрезентативными; в последнем азиридин образует на месте и открывается через нуклеофильная атака сформировать соответствующий амин.[3][8]

Синтез циклопропанов
Для добавления сернистых илидов к енонам выше 1,4-селективность обычно получают с сульфоксониевыми реагентами, чем с сульфониевыми реагентами. Было показано, что многие электроноакцепторные группы совместимы с реакцией, включая кетоны, сложные эфиры, и амиды (пример ниже включает Вайнреб амид ). В других сопряженных системах 1,6-присоединение имеет тенденцию преобладать над 1,4-присоединением.[3][8]

Другие реакции
В дополнение к реакциям, первоначально описанным Джонсоном, Кори и Чайковским, серные илиды использовались для ряда связанных реакции омологации которые обычно группируются под одним и тем же именем.
- С участием эпоксиды и азиридины реакция служит расширением кольца с образованием соответствующего оксетан или азетидин. Длительное время реакции, необходимое для этих реакций, предотвращает их протекание в столь значительной степени. побочные реакции при синтезе эпоксидов и азиридинов.[8]

- Несколько циклоприсоединения где илид служит "нуклеофильный карбеноид эквивалент ».[8]
![[4 + 1] циклоприсоединение с реактивом Кори – Чайковского.](http://upload.wikimedia.org/wikipedia/commons/thumb/c/cc/CCR41.png/320px-CCR41.png)
- Живая полимеризация с помощью триалкилбораны в качестве катализатора и (диметилоксосульфанил) метанид в качестве мономера были описаны для синтеза различных сложных полимеров.[12]

Энантиоселективные варианты
Развитие энантиоселективный (т. е. получение энантиомерный избыток (обозначается как «ее») вариант реакции Джонсона – Кори – Чайковского остается активной областью академических исследований. Использование хиральный сульфиды в стехиометрический мода оказалась более успешной, чем соответствующая каталитический варианты, но объем подложки по-прежнему ограничен во всех случаях. Каталитические варианты были разработаны почти исключительно для энантиоселективных целей; типичные органосульфидные реагенты не являются чрезмерно дорогими, и рацемические реакции могут проводиться с эквимолярными количествами илида без значительного увеличения затрат. С другой стороны, хиральные сульфиды дороже в получении, что способствует развитию каталитических энантиоселективных методов.[2]
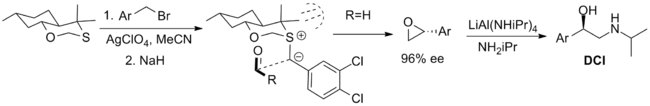
Стехиометрические реагенты
Наиболее успешные реагенты, используемые в стехиометрическом порядке, показаны ниже. Первый - это бициклический оксатиан, который был использован в синтезе β-адренергического соединения дихлоризопротеренол (DCI), но ограничен доступностью только одного энантиомера реагента. Синтез осевой диастереомер рационализируется через 1,3-аномерный эффект что снижает нуклеофильность экваториальный одинокая пара. В конформация илиды ограничено трансаннулярная деформация и приближение альдегида ограничено одной стороной илида стерическими взаимодействиями с метильными заместителями.[2]
Другой важный реагент - это камфора -производный реагент, разработанный Вариндер Аггарвал из Бристольский университет. И то и другое энантиомеры легко синтезируются, хотя выходы ниже, чем для оксатианового реагента. Конформация илида определяется взаимодействием с плацдарм водород и приближение альдегида блокируется камфарой часть. В реакции используется фосфазен основа, способствующая образованию илида.[2]
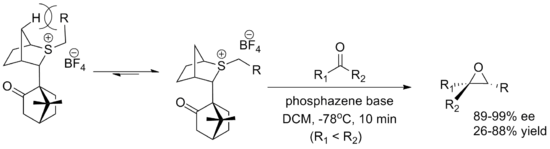
Каталитические реагенты
Каталитические реагенты оказались менее успешными, при этом большинство вариантов страдали низким выходом, низкой энантиоселективностью или тем и другим. Также существуют проблемы с объемом субстрата, большинство из которых имеют ограничения на перенос метилена и алифатический альдегиды. Проблема связана с необходимостью нуклеофильный сульфид, который эффективно генерирует илид, который также может действовать как хороший уходящая группа с образованием эпоксида. Поскольку факторы, лежащие в основе этих желаний, противоречат друг другу, настройка свойств катализатора оказалась сложной. Ниже показаны некоторые из наиболее успешных катализаторов, а также выходы и энантиомерный избыток для их использования в синтезе (E) -стильбен окись.[2]

Аггарвал разработал альтернативный метод, в котором используется тот же сульфид, что и выше, и новое алкилирование, включающее родий карбеноид сформированный на месте. Этот метод также имеет ограниченную область применения субстрата, он не работает ни при каких условиях. электрофилы обладающие основными заместителями за счет конкурентоспособное потребление карбеноида.[2]

Смотрите также
использованная литература
- ^ а б c d е ж г Aggarwal, V.K .; Ричардсон, Дж. (2003). «Сложность катализа: истоки энантио- и диастереоконтроля в реакциях эпоксидирования, опосредованных сернистым илидом». Химические коммуникации (21): 2644. Дои:10.1039 / b304625g. PMID 14649793.
- ^ а б c d е ж Aggarwal, V.K .; Винн, К. Л. (2004). «Каталитическое, асимметричное эпоксидирование с помощью илида серы карбонильных соединений: объем, селективность и применение в синтезе». Отчеты о химических исследованиях. 37 (8): 611–620. Дои:10.1021 / ar030045f. PMID 15311960.
- ^ а б c Гололобов Ю.Г .; Несмеянов, А. Н .; лысенко, В.П .; Болдескул, И. Э. (1987). «Двадцать пять лет этилида диметилсульфоксония (реактив Кори)». Тетраэдр. 43 (12): 2609–2651. Дои:10.1016 / s0040-4020 (01) 86869-1.
- ^ а б Li, A.-H .; Dai, L.-X .; Аггарвал В. К. (1997). «Асимметричные реакции илида: эпоксидирование, циклопропанирование, азиридинирование, олефинирование и перегруппировка». Химические обзоры. 97 (6): 2341–2372. Дои:10.1021 / cr960411r. PMID 11848902.
- ^ McGarrigle, E.M .; Myers, E. L .; Illa, O .; Shaw, M. A .; Riches, S. L .; Аггарвал В. К. (2007). «Халькогениды как органокатализаторы». Химические обзоры. 107 (12): 5841–5883. Дои:10.1021 / cr068402y. PMID 18072810.
- ^ Johnson, A.W .; ЛаКонт, Р. Б. (1961). «Химия Илидов. VI. Флуоренилид диметилсульфония - синтез эпоксидов». Варенье. Chem. Soc. 83 (2): 417–423. Дои:10.1021 / ja01463a040.
- ^ Кори, Э. Дж.; Чайковский, М. (1965). "Метилид диметилоксосульфония ((CH3)2СОЧ2) и метилид диметилсульфония ((CH3)2SCH2). Образование и применение в органическом синтезе ». Варенье. Chem. Soc. 87 (6): 1353–1364. Дои:10.1021 / ja01084a034.
- ^ а б c d е Ли, Джек Джи (2005). Именованные реакции в химии гетероциклов. Хобокен, Нью-Джерси: John Wiley & Sons, Inc., стр. 2–14. ISBN 9780471704140.
- ^ Mundy, Bradford, P .; Эллерд, Майкл Д .; Фавалоро, Фрэнк Г. мл. (2005). Назовите реакции и реагенты в органической химии (2-е изд.). Хобокен, Нью-Джерси: John Wiley & Sons, Inc., стр. 174–175, 743. ISBN 9780471739869.CS1 maint: несколько имен: список авторов (ссылка на сайт)
- ^ Данишефский, С. Дж .; Мастерс, Дж. Дж .; Young, W. B .; Link, J. T .; Снайдер, Л. Б .; Magee, T. V .; Юнг, Д. К .; Isaacs, R.CA .; Bornmann, W. G .; Alaimo, C.A .; Coburn, C.A .; Ди Гранди, М. Дж. (1996). «Полный синтез баккатина III и таксола». Журнал Американского химического общества. 118 (12): 2843–2859. Дои:10.1021 / ja952692a.
- ^ Kuehne, M.E .; Сюй Ф. (1993). «Полный синтез алкалоидов стрихнана и аспидосперматана. 3. Полный синтез (. + -.) - стрихнина». Журнал органической химии. 58 (26): 7490–7497. Дои:10.1021 / jo00078a030.
- ^ Luo, J .; Ши, К. Дж. (2010). «Полигомологация. Живая полимеризация С1». Отчеты о химических исследованиях. 43 (11): 1420–1433. Дои:10.1021 / ar100062a. PMID 20825177.
{kind=link}