Структурное выравнивание - Structural alignment
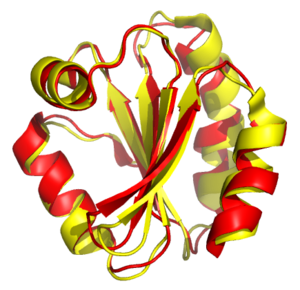
Структурное выравнивание попытки установить гомология между двумя или более полимер конструкции на основе их формы и трехмерности конформация. Этот процесс обычно применяется к белок третичные структуры но также может использоваться для больших РНК молекулы. В отличие от простой структурной суперпозиции, где известны по крайней мере некоторые эквивалентные остатки двух структур, структурное выравнивание не требует априори знание равнозначных позиций. Структурное выравнивание - ценный инструмент для сравнения белков с низким сходством последовательностей, где эволюционные отношения между белками не могут быть легко обнаружены стандартными методами. выравнивание последовательностей техники. Поэтому структурное выравнивание можно использовать для обозначения эволюционный отношения между белками, которые имеют очень мало общей последовательности. Однако следует проявлять осторожность при использовании результатов в качестве доказательства общего эволюционного происхождения из-за возможных смешивающих эффектов конвергентная эволюция посредством чего несколько не связанных между собой аминокислота последовательности сходятся на общем третичная структура.
Структурные выравнивания могут сравнивать две последовательности или несколько последовательностей. Поскольку эти сопоставления основываются на информации о трехмерных конформациях всех запрашиваемых последовательностей, метод может использоваться только для последовательностей, для которых эти структуры известны. Обычно их находят Рентгеновская кристаллография или же ЯМР-спектроскопия. Возможно выполнение структурного выравнивания конструкций, изготовленных предсказание структуры методы. Действительно, оценка таких прогнозов часто требует структурного согласования между моделью и реально известной структурой для оценки качества модели.[1] Структурное выравнивание особенно полезно при анализе данных из структурная геномика и протеомика усилий, и их можно использовать в качестве точек сравнения для оценки выравниваний, произведенных чисто последовательностями биоинформатика методы.[2][3][4]
Выходы структурного выравнивания представляют собой суперпозицию атомных наборы координат и минимальный среднеквадратическое значение отклонение (RMSD ) между конструкциями. RMSD двух выровненных структур указывает на их расхождение друг с другом. Структурное выравнивание может быть затруднено наличием нескольких белковые домены внутри одной или нескольких входных структур, поскольку изменения относительной ориентации доменов между двумя выравниваемыми структурами могут искусственно завышать RMSD.
Данные, полученные в результате структурного выравнивания
Минимальная информация, полученная в результате успешного структурного выравнивания, - это набор остатков, которые считаются эквивалентными между структурами. Этот набор эквивалентностей затем обычно используется для наложения трехмерных координат для каждой входной структуры. (Обратите внимание, что один входной элемент может быть зафиксирован в качестве эталона, и поэтому его наложенные координаты не изменяются.) Подобранные структуры могут использоваться для расчета взаимных значений RMSD, а также других более сложных мер структурного сходства, таких как глобальный тест расстояния (GDT,[5] метрика, используемая в CASP ). Структурное выравнивание также подразумевает соответствующее одномерное выравнивание последовательностей из которых идентичность последовательностей или процент остатков, которые идентичны между входными структурами, могут быть рассчитаны как мера того, насколько тесно связаны две последовательности.
Типы сравнений
Поскольку белковые структуры состоят из аминокислоты чей боковые цепи связаны общим белковым каркасом, ряд различных возможных подмножеств атомов, составляющих макромолекулу белка, можно использовать для получения структурного выравнивания и вычисления соответствующих значений RMSD. При выравнивании структур с очень разными последовательностями атомы боковой цепи обычно не принимаются во внимание, поскольку их идентичность различается между многими выровненными остатками. По этой причине методы структурного выравнивания обычно используют по умолчанию только атомы основной цепи, включенные в пептидная связь. Для простоты и эффективности часто только альфа-углерод позиции учитываются, так как пептидная связь имеет минимально вариант планарный конформация. Только когда выравниваемые структуры очень похожи или даже идентичны, имеет смысл выровнять положения атомов боковой цепи, и в этом случае RMSD отражает не только конформацию остова белка, но также и ротамерный состояния боковых цепей. Другие критерии сравнения, снижающие шум и поддерживающие положительные совпадения, включают: вторичная структура назначение, родной контакт карты или паттерны взаимодействия остатков, меры упаковки боковой цепи и меры водородная связь удержание.[6]
Структурная суперпозиция
Самое простое возможное сравнение белковых структур не пытается выровнять входные структуры и требует предварительно рассчитанного выравнивания в качестве входных данных, чтобы определить, какие остатки в последовательности должны учитываться при вычислении RMSD. Структурная суперпозиция обычно используется для сравнения нескольких конформаций одного и того же белка (в этом случае выравнивание не требуется, поскольку последовательности одинаковы) и для оценки качества выравниваний, произведенных с использованием только информации о последовательностях между двумя или более последовательностями, структура которых известна. . В этом методе традиционно используется простой алгоритм аппроксимации методом наименьших квадратов, в котором оптимальные повороты и перемещения находятся путем минимизации суммы квадратов расстояний между всеми структурами в суперпозиции.[7] В последнее время методы максимального правдоподобия и байесовские методы значительно повысили точность расчетных матриц вращения, сдвигов и ковариаций для суперпозиции.[8][9]
Алгоритмы, основанные на многомерных поворотах и модифицированные кватернионы были разработаны для идентификации топологических отношений между белковыми структурами без необходимости предварительно заданного выравнивания. Такие алгоритмы успешно идентифицировали канонические свертки, такие как четырехспиральный пучок.[10] В SuperPose метод достаточно расширяемый, чтобы исправить относительные повороты доменов и другие структурные ошибки.[11]
Оценка сходства
Часто целью поиска структурной суперпозиции является не столько сама суперпозиция, сколько оценка сходства двух структур или уверенность в отдаленном совмещении.[1][2][3] Тонкое, но важное отличие от максимальной структурной суперпозиции - преобразование выравнивания в значимый показатель сходства.[12][13] Большинство методов выводят своего рода «оценку», указывающую качество наложения.[5] [14] [15][12][13] Однако на самом деле хочется нет просто по оценкам «Z-оценка» или по оценкам Е-ценность случайного наблюдения наблюдаемой суперпозиции, но вместо этого желательно, чтобы по оценкам E-value - это тесная корреляция с истинным E-value. Критично, даже если оценочное значение E метода точно верно в среднем, если ему не хватает низкого стандартного отклонения в процессе генерации оценочного значения, то порядок ранжирования относительных сходств белка запроса и сравнительного набора редко будет соответствовать «истинному» порядку.[12][13]
Различные методы будут накладывать разное количество остатков, потому что они используют разные гарантии качества и разные определения «перекрытия»; некоторые включают только остатки, отвечающие множеству локальных и глобальных критериев суперпозиции, а другие являются более жадными, гибкими и беспорядочными. Большее количество наложенных друг на друга атомов может означать большее сходство, но это не всегда может давать лучшее E-значение, количественно определяющее маловероятность суперпозиции, и, следовательно, не так полезно для оценки сходства, особенно в удаленных гомологах.[1][2][3][4]
Алгоритмическая сложность
Оптимальное решение
Оптимальный "заправка "белковой последовательности на известную структуру, и было показано, что получение оптимального множественного выравнивания последовательностей НП-полный.[16][17] Однако это не означает, что проблема структурного выравнивания является NP-полной. Строго говоря, оптимальное решение проблемы выравнивания структуры белка известно только для определенных мер сходства структуры белка, таких как меры, используемые в экспериментах по предсказанию структуры белка, GDT_TS[5] и MaxSub.[14] Эти меры могут быть строго оптимизированы с помощью алгоритма, способного максимизировать количество атомов в двух белках, которые могут быть наложены друг на друга при заранее заданном отрезке расстояния.[15] К сожалению, алгоритм оптимального решения непрактичен, поскольку время его работы зависит не только от длины, но и от внутренней геометрии входных белков.
Примерное решение
Приблизительно полиномиальное время были разработаны алгоритмы структурного выравнивания, которые производят семейство «оптимальных» решений в пределах параметра приближения для данной функции оценки.[15][18] Хотя эти алгоритмы теоретически классифицируют проблему приблизительного выравнивания структуры белка как «решаемую», они по-прежнему слишком дороги в вычислительном отношении для крупномасштабного анализа структуры белка. Как следствие, практических алгоритмов, которые сходятся к глобальным решениям выравнивания с учетом функции оценки, не существует. Таким образом, большинство алгоритмов являются эвристическими, но были разработаны алгоритмы, которые гарантируют сходимость, по крайней мере, к локальным максимизаторам функций подсчета и являются практичными.[19]
Представление структур
Белковые структуры должны быть представлены в некотором координатно-независимом пространстве, чтобы сделать их сопоставимыми. Обычно это достигается путем построения матрицы от последовательности к последовательности или серии матриц, которые включают сравнительные показатели, а не абсолютные расстояния относительно фиксированного координатного пространства. Интуитивное представление - это матрица расстояний, который является двумерным матрица содержащие все попарные расстояния между некоторым подмножеством атомов в каждой структуре (например, альфа-углерод ). Размерность матрицы увеличивается по мере увеличения количества одновременно выравниваемых структур. Уменьшение количества протеина до грубого показателя, такого как вторичная структура элементы (SSE) или структурные фрагменты также могут производить разумное выравнивание, несмотря на потерю информации из-за отбрасывания расстояний, поскольку шум также отбрасывается.[20] Выбор представления для облегчения вычислений имеет решающее значение для разработки эффективного механизма выравнивания.
Методы
Методы структурного выравнивания использовались при сравнении отдельных структур или наборов структур, а также при создании баз данных сравнения «все ко всем», которые измеряют расхождение между каждой парой структур, присутствующих в Банк данных белков (PDB). Такие базы данных используются для классификации белков по их складывать.
ДАЛИ

Распространенным и популярным методом структурного выравнивания является DALI, или метод ALIgnment матрицы расстояний, который разбивает входные структуры на гексапептидные фрагменты и вычисляет матрицу расстояний, оценивая образцы контактов между последовательными фрагментами.[21] Вторичная структура признаки, которые включают остатки, которые являются смежными в последовательности, появляются на матрице главная диагональ; другие диагонали в матрице отражают пространственные контакты между остатками, которые не находятся рядом друг с другом в последовательности. Когда эти диагонали параллельны главной диагонали, элементы, которые они представляют, параллельны; когда они перпендикулярны, их черты антипараллельны. Это представление требует большого объема памяти, поскольку элементы квадратной матрицы симметричны (и, следовательно, избыточны) относительно главной диагонали.
Когда матрицы расстояний двух белков имеют одинаковые или похожие характеристики в примерно одинаковых положениях, можно сказать, что они имеют похожие складки с петлями одинаковой длины, соединяющими их элементы вторичной структуры. Фактический процесс выравнивания DALI требует поиска сходства после построения матриц расстояний двух белков; обычно это выполняется через серию перекрывающихся подматриц размером 6x6. Затем совпадения подматриц повторно собираются в окончательное выравнивание с помощью стандартного алгоритма максимизации очков - в исходной версии DALI использовалась Монте-Карло моделирование, чтобы максимизировать показатель структурного сходства, который является функцией расстояний между предполагаемыми соответствующими атомами. В частности, более удаленные атомы в пределах соответствующих элементов экспоненциально уменьшаются, чтобы уменьшить влияние шума, вносимого подвижностью петель, скручиванием спирали и другими незначительными структурными изменениями.[20] Поскольку DALI полагается на матрицу расстояний «все ко всем», он может учитывать возможность того, что структурно выровненные объекты могут появляться в разных порядках в двух сравниваемых последовательностях.
Метод DALI также использовался для создания базы данных, известной как ФССП (Классификация складок, основанная на выравнивании структуры и структуры белков или семейств структурно похожих белков), в которой все известные белковые структуры выравниваются друг с другом для определения их структурных соседей и классификации складок. Существует база данных с возможностью поиска на основе DALI, а также загружаемая программа и веб-поиск основан на отдельной версии, известной как DaliLite.
Комбинаторное расширение
Метод комбинаторного расширения (CE) похож на DALI в том, что он также разбивает каждую структуру в наборе запросов на серию фрагментов, которые затем пытается повторно собрать в полное выравнивание. Серия попарных комбинаций фрагментов, называемых парами выровненных фрагментов или AFP, используется для определения матрицы сходства, с помощью которой генерируется оптимальный путь для идентификации окончательного совмещения. Только AFP, которые соответствуют заданным критериям локального сходства, включаются в матрицу как средство уменьшения необходимого пространства поиска и, таким образом, повышения эффективности.[22] Возможен ряд показателей сходства; первоначальное определение метода CE включало только структурные суперпозиции и расстояния между остатками, но с тех пор было расширено за счет включения местных экологических свойств, таких как вторичная структура, воздействие растворителя, образцы водородных связей и двугранные углы.[22]
Путь выравнивания рассчитывается как оптимальный путь через матрицу сходства путем линейного продвижения по последовательностям и расширения выравнивания со следующей возможной парой AFP с высоким баллом. Начальная пара AFP, которая является ядром выравнивания, может находиться в любой точке матрицы последовательностей. Затем расширения переходят к следующему AFP, который соответствует заданным критериям расстояния, ограничивая выравнивание небольшими размерами зазора. Размер каждого AFP и максимальный размер зазора являются обязательными входными параметрами, но обычно устанавливаются на эмпирически определенные значения 8 и 30 соответственно.[22] Подобно DALI и SSAP, CE использовался для построения всеобъемлющей классификации база данных из известных белковых структур в PDB.
В RCSB PDB недавно выпустила обновленную версию CE, Mammoth и FATCAT как часть Инструмент сравнения белков RCSB PDB. Он предоставляет новый вариант CE, который может обнаруживать круговые перестановки в белковых структурах.[23]
Мамонт
МАМОНТ [12] подходит к проблеме согласования с другой цели, чем почти все другие методы. Вместо того, чтобы пытаться найти выравнивание, которое максимально перекрывает наибольшее количество остатков, он ищет подмножество структурного выравнивания, которое с наименьшей вероятностью произойдет случайно. Для этого он отмечает локальное выравнивание мотива флажками, чтобы указать, какие остатки одновременно удовлетворяют более строгим критериям: 1) перекрытие локальных структур 2) регулярная вторичная структура 3) 3D-суперпозиция 4) такой же порядок в первичной последовательности. Он преобразует статистику количества остатков с совпадениями с высокой степенью достоверности и размера белка, чтобы вычислить значение ожидания для случайного результата. Он превосходно подходит для сопоставления удаленных гомологов, особенно структур, созданных с помощью предсказания структуры ab initio, с семействами структур, такими как SCOP, потому что он подчеркивает извлечение статистически надежного суб-выравнивания, а не достижение максимального выравнивания последовательностей или максимальной трехмерной суперпозиции.[2][3]
Для каждого перекрывающегося окна из 7 последовательных остатков он вычисляет набор единичных векторов направления смещения между соседними остатками C-альфа. Локальные мотивы «все против всех» сравниваются на основе оценки URMS. Эти значения становятся записями оценки парного выравнивания для динамического программирования, которое производит начальное попарное выравнивание остатков. На втором этапе используется модифицированный алгоритм MaxSub: одна 7-местная выровненная пара в каждом белке используется для ориентации двух полноразмерных белковых структур, чтобы максимально наложить их только на эти 7 C-альфа, затем в этой ориентации он сканирует любые дополнительные выровненные пары близкие в 3D. Он переориентирует структуры, чтобы наложить этот расширенный набор, и выполняет итерацию до тех пор, пока не перестанут совпадать пары в 3D. Этот процесс перезапускается для каждых 7 окон остатков в выравнивании семян. Результатом является максимальное количество атомов, найденное из любого из этих начальных семян. Эта статистика преобразуется в откалиброванное значение E для сходства белков.
Mammoth не пытается повторить первоначальное выравнивание или расширить подмножество высокого качества. Поэтому отображаемое начальное выравнивание нельзя справедливо сравнивать с выравниванием DALI или TM, поскольку оно было сформировано просто как эвристика для сокращения пространства поиска. (Его можно использовать, если нужно выравнивание, основанное исключительно на локальном сходстве структуры и мотива, независимо от атомного выравнивания твердого тела на больших расстояниях.) Из-за той же экономичности он работает более чем в десять раз быстрее, чем DALI, CE и TM-align. [24]Он часто используется в сочетании с этими более медленными инструментами для предварительного просмотра больших баз данных для извлечения только лучших структур, связанных с E-значением, для более исчерпывающего наложения или дорогостоящих вычислений. [25][26]
Он оказался особенно успешным при анализе структур-ловушек на основе предсказания структуры ab initio.[1][2][3] Эти приманки печально известны тем, что получают правильную структуру мотивов локальных фрагментов и формируют некоторые ядра правильной трехмерной третичной структуры, но получают неправильную третичную структуру полной длины. В этом сумеречном режиме удаленной гомологии е-значения Мамонта для CASP[1] Было показано, что оценка предсказания структуры белка значительно больше коррелирует с рейтингом человека, чем SSAP или DALI.[12] Способность мамонтов извлекать многокритериальные частичные перекрытия с белками известной структуры и ранжировать их по правильным E-значениям в сочетании с его скоростью облегчает сканирование огромного количества моделей-приманок по базе данных PDB для определения наиболее вероятных правильных приманок на основе их удаленная гомология с известными белками. [2]
SSAP
Метод SSAP (программа последовательного выравнивания структуры) использует двойное динамическое программирование произвести структурное выравнивание на основе атома к атому векторов в пространстве структуры. Вместо альфа-углеродов, обычно используемых при структурном выравнивании, SSAP конструирует свои векторы из бета-углерод для всех остатков, кроме глицина, метод, который, таким образом, учитывает ротамерное состояние каждого остатка, а также его расположение вдоль основной цепи. SSAP сначала создает серию векторов расстояний между остатками между каждым остатком и его ближайшими несмежными соседями на каждом белке. Затем строится серия матриц, содержащих разности векторов между соседями для каждой пары остатков, для которых были построены векторы. Динамическое программирование, применяемое к каждой результирующей матрице, определяет ряд оптимальных локальных выравниваний, которые затем суммируются в «итоговую» матрицу, к которой снова применяется динамическое программирование для определения общего структурного выравнивания.
Первоначально SSAP производил только попарные выравнивания, но с тех пор был расширен и на множественные выравнивания.[27] Он был применен универсально для создания иерархической схемы классификации складок, известной как CATH (Класс, Архитектура, Топология, Гомология),[28] который был использован для построения Классификация структуры белка CATH база данных.
Последние достижения
Улучшения в методах структурного выравнивания составляют активную область исследований, и часто предлагаются новые или модифицированные методы, которые, как утверждается, предлагают преимущества по сравнению с более старыми и более широко распространенными методами. Недавний пример, TM-align, использует новый метод взвешивания своей матрицы расстояний, к которому стандарт динамическое программирование затем применяется.[29][13] Взвешивание предлагается для ускорения сходимости динамического программирования и корректировки эффектов, возникающих из-за длин выравнивания. В ходе сравнительного исследования сообщалось, что TM-align улучшает скорость и точность по сравнению с DALI и CE.[29]
Другими многообещающими методами структурного выравнивания являются методы местного структурного выравнивания. Они обеспечивают сравнение предварительно выбранных частей белков (например, сайтов связывания, определяемых пользователем структурных мотивов) [30][31][32] против сайтов связывания или структурных баз данных цельного белка. Серверы MultiBind и MAPPIS [32][33] позволяют идентифицировать общее пространственное расположение физико-химических свойств, таких как донор водородной связи, акцептор, алифатические, ароматические или гидрофобные, в наборе предоставленных пользователем сайтов связывания белков, определяемых взаимодействиями с небольшими молекулами (MultiBind) или в наборе предоставленных пользователем белок-белковые интерфейсы (MAPPIS). Другие обеспечивают сравнение целых белковых структур. [34] против ряда представленных пользователем структур или против большой базы данных структур белков в разумные сроки (ProBiS[35]). В отличие от подходов глобального выравнивания, подходы локального структурного выравнивания подходят для обнаружения локально консервативных паттернов функциональных групп, которые часто появляются в сайтах связывания и имеют значительное участие в связывании лиганда.[33] В качестве примера, сравнивая G-Losa,[36] инструмент для локального выравнивания структуры с TM-align, методом на основе глобального выравнивания структуры. В то время как G-Losa предсказывает положения лекарственно-подобных лигандов в одноцепочечных белковых мишенях более точно, чем TM-align, общая вероятность успеха TM-align выше.[37]
Однако по мере того, как улучшения алгоритмов и производительность компьютеров устранили чисто технические недостатки старых подходов, стало ясно, что не существует единого универсального критерия для «оптимального» структурного согласования. TM-align, например, особенно надежен при количественной оценке сравнений между наборами белков с большими различиями в длинах последовательностей, но он только косвенно фиксирует водородные связи или сохранение порядка вторичной структуры, что может быть лучшим показателем для выравнивания эволюционно связанных белков. Таким образом, последние разработки были сосредоточены на оптимизации определенных атрибутов, таких как скорость, количественная оценка, корреляция с альтернативными золотыми стандартами или допустимость несовершенства структурных данных или структурных моделей ab initio. Альтернативная методология, которая набирает популярность, заключается в использовании консенсус различных методов для установления структурного сходства белков.[38]
Эта секция нуждается в расширении с: добавить обсуждение следующих тем: A) гибкое выравнивание по сравнению с твердым телом B) зависимое от порядка последовательности по сравнению с независимым C) выравнивание биологических сборок[39]. Вы можете помочь добавляя к этому. (Июль 2012 г.) |
Структурное выравнивание РНК
Методы структурного выравнивания традиционно применялись исключительно к белкам в качестве основных биологических макромолекулы которые предполагают характерные трехмерные структуры. Однако большие РНК молекулы также образуют характерные третичные структуры, которые в первую очередь опосредуются водородные связи сформированный между пар оснований а также базовая укладка. Функционально похожий некодирующая РНК молекулы может быть особенно трудно извлечь из геномика данные, потому что структура более консервативна, чем последовательность в РНК, а также в белках,[40] а более ограниченный алфавит РНК снижает информационное содержание любого данного нуклеотид в любой позиции.
Однако из-за растущего интереса к структурам РНК и из-за роста числа экспериментально определенных структур 3D РНК, в последнее время было разработано мало методов подобия структур РНК. Один из таких методов, например, SETTER[41] который разбивает каждую структуру РНК на более мелкие части, называемые общими вторичными структурными единицами (GSSU). Затем GSSU выравниваются, и эти частичные выравнивания объединяются в окончательное выравнивание структуры РНК и оцениваются. Метод реализован в SETTER веб-сервер.[42]
Недавно опубликован и реализован в программе метод попарного структурного выравнивания последовательностей РНК с низкой идентичностью последовательностей. FOLDALIGN.[43] Однако этот метод не совсем аналогичен методам структурного выравнивания белков, потому что он с помощью вычислений предсказывает структуры входных последовательностей РНК, а не требует экспериментально определенных структур в качестве входных данных. Хотя вычислительное предсказание сворачивание белка на сегодняшний день не был особенно успешным, структуры РНК без псевдоузлы часто можно разумно предсказать, используя свободная энергия методы подсчета очков, учитывающие спаривание и укладку оснований.[44]
Программного обеспечения
Выбор программного инструмента для структурного выравнивания может быть сложной задачей из-за большого разнообразия доступных пакетов, которые значительно различаются по методологии и надежности. Частичное решение этой проблемы было представлено в [38] и сделаны общедоступными через веб-сервер ProCKSI. Более полный список доступного в настоящее время и свободно распространяемого программного обеспечения для выравнивания конструкций можно найти в программное обеспечение для выравнивания конструкций.
Свойства некоторых серверов структурного выравнивания и пакетов программного обеспечения обобщены и протестированы с примерами на Инструменты структурного выравнивания в Proteopedia.Org.
Смотрите также
Рекомендации
- ^ а б c d е Крыштафович А, Монастырский Б, Фиделис К. (2016). «Статистика CASP11 и система оценки центра прогнозов». Белки. 84: (Приложение 1): 15-19. Дои:10.1002 / prot.25005.CS1 maint: использует параметр авторов (связь)
- ^ а б c d е ж Ларс Мальмстрем, Майкл Риффл, Чарли Э.М. Штраус, Дилан Чивиан, Триша Н. Дэвис, Ричард Бонно, Дэвид Бейкер (2007). «Присвоение надсемейству протеома дрожжей посредством интеграции предсказания структуры с генной онтологией». PLoS Biol. 5 (4): e76соответствующий автор1, 2. Дои:10.1371 / journal.pbio.0050076.CS1 maint: использует параметр авторов (связь)
- ^ а б c d е Дэвид Э. Ким, Дилан Чивиан и Дэвид Бейкер (2004). «Прогнозирование и анализ структуры белка с помощью сервера Robetta». Исследования нуклеиновых кислот. 32 (выпуск веб-сервера): W526 – W531. Дои:10.1093 / нар / гх468. PMID 15215442.CS1 maint: использует параметр авторов (связь)
- ^ а б Чжан И, Сколник Дж (2005). «Проблема предсказания структуры белка может быть решена с использованием текущей библиотеки PDB». Proc Natl Acad Sci USA. 102 (4): 1029–34. Дои:10.1073 / pnas.0407152101. ЧВК 545829. PMID 15653774.
- ^ а б c Земля А. (2003). «LGA - метод обнаружения трехмерных сходств в структурах белков». Исследования нуклеиновых кислот. 31 (13): 3370–3374. Дои:10.1093 / нар / gkg571. ЧВК 168977. PMID 12824330.
- ^ Годзик А (1996). «Структурное выравнивание двух белков: есть ли уникальный ответ?». Белковая наука. 5 (7): 1325–38. Дои:10.1002 / pro.5560050711. ЧВК 2143456. PMID 8819165.
- ^ Мартин ACR (1982). «Быстрое сравнение белковых структур». Acta Crystallogr A. 38 (6): 871–873. Дои:10.1107 / S0567739482001806.
- ^ Теобальд Д.Л., Вуттке Д.С. (2006). «Эмпирические байесовские иерархические модели для регуляризации оценки максимального правдоподобия в матричной задаче Гаусса Прокруста». Труды Национальной академии наук. 103 (49): 18521–18527. Дои:10.1073 / pnas.0508445103. ЧВК 1664551. PMID 17130458.
- ^ Теобальд Д.Л., Вуттке Д.С. (2006). «THESEUS: Максимальное правдоподобие наложения и анализ макромолекулярных структур». Биоинформатика. 22 (17): 2171–2172. Дои:10.1093 / биоинформатика / btl332. ЧВК 2584349. PMID 16777907.
- ^ Дидерикс К. (1995). «Структурная суперпозиция белков с неизвестным выравниванием и обнаружение топологического сходства с использованием алгоритма шестимерного поиска». Белки. 23 (2): 187–95. Дои:10.1002 / prot.340230208. PMID 8592700.
- ^ Маити Р., Ван Домселаар Г. Х., Чжан Х., Вишарт Д. С. (2004). «SuperPose: простой сервер для сложной структурной суперпозиции». Нуклеиновые кислоты Res. 32 (Выпуск веб-сервера): W590–4. Дои:10.1093 / нар / гх477. ЧВК 441615. PMID 15215457.
- ^ а б c d е Ортис, АР; Штраус CE; Ольмеа О. (2002). «МАМОНТ (сопоставление молекулярных моделей, полученных из теории): автоматизированный метод сравнения моделей». Белковая наука. 11 (11): 2606–2621. Дои:10.1110 / пс. 0215902. PMID 12381844.
- ^ а б c d Чжан И, Сколник Дж (2004). «Функция скоринга для автоматической оценки качества шаблона структуры белка». Белки. 57 (4): 702–710. Дои:10.1002 / prot.20264. PMID 15476259.
- ^ а б Сью Н., Элофссон А., Рихлевск Л., Фишер Д. (2000). «MaxSub: автоматизированная мера для оценки качества предсказания структуры белка». Биоинформатика. 16 (9): 776–85. Дои:10.1093 / биоинформатика / 16.9.776. PMID 11108700.
- ^ а б c Полексич А (2009). «Алгоритмы оптимального выравнивания структуры белка». Биоинформатика. 25 (21): 2751–2756. Дои:10.1093 / биоинформатика / btp530. PMID 19734152.
- ^ Lathrop RH. (1994). «Проблема потоковой передачи белков с предпочтениями взаимодействия аминокислотных последовательностей является NP-полной». Protein Eng. 7 (9): 1059–68. CiteSeerX 10.1.1.367.9081. Дои:10.1093 / белок / 7.9.1059. PMID 7831276.
- ^ Ван Л., Цзян Т. (1994). «О сложности множественного выравнивания последовательностей». Журнал вычислительной биологии. 1 (4): 337–48. CiteSeerX 10.1.1.408.894. Дои:10.1089 / cmb.1994.1.337. PMID 8790475.
- ^ Колодный Р, Линиал Н (2004). «Приблизительное структурное выравнивание белка за полиномиальное время». PNAS. 101 (33): 12201–12206. Дои:10.1073 / pnas.0404383101. ЧВК 514457. PMID 15304646.
- ^ Мартинес Л., Андреани, Р., Мартинес, Дж. М.. (2007). «Конвергентные алгоритмы структурного выравнивания белков». BMC Bioinformatics. 8: 306. Дои:10.1186/1471-2105-8-306. ЧВК 1995224. PMID 17714583.CS1 maint: несколько имен: список авторов (связь)
- ^ а б Крепление DM. (2004). Биоинформатика: анализ последовательности и генома 2-е изд. Лабораторный пресс Колд-Спринг-Харбор: Колд-Спринг-Харбор, Нью-Йорк ISBN 0879697121
- ^ Холм Л., Сандер С. (1996). «Картирование белковой вселенной». Наука. 273 (5275): 595–603. Дои:10.1126 / science.273.5275.595. PMID 8662544.
- ^ а б c Шиндялов, И.Н .; Bourne P.E. (1998). «Выравнивание структуры белка путем возрастающего комбинаторного удлинения (CE) оптимального пути». Белковая инженерия. 11 (9): 739–747. Дои:10.1093 / белок / 11.9.739. PMID 9796821.
- ^ Prlic A, Bliven S, Rose PW, Bluhm WF, Bizon C, Godzik A, Bourne PE (2010). «Предварительно рассчитанные выравнивания структуры белков на сайте RCSB PDB». Биоинформатика. 26 (23): 2983–2985. Дои:10.1093 / биоинформатика / btq572. ЧВК 3003546. PMID 20937596.
- ^ Пин-Хао Чи, Бин Панг, Дмитрий Коркин, Чи-Рен Шю (2009). «Эффективная SCOP-кратная классификация и извлечение с использованием сопоставления субструктур белков на основе индексов». Биоинформатика. 25 (19): 2559–2565. Дои:10.1093 / биоинформатика / btp474.CS1 maint: использует параметр авторов (связь)
- ^ Сара Чик, Юань Ци, Шри Кришна, Лиза Н. Кинч и Ник В. Гришин (2004). «SCOPmap: автоматическое отнесение белковых структур к эволюционным суперсемействам». BMC Bioinformatics. 5 (197). Дои:10.1186/1471-2105-5-197. PMID 15598351.CS1 maint: использует параметр авторов (связь)
- ^ Кай Ван, Рам Самудрала. «FSSA: новый метод определения функциональных сигнатур по структурным согласованиям». Биоинформатика. 21 (13): 2969–2977. Дои:10.1093 / биоинформатика / bti471.CS1 maint: использует параметр авторов (связь)
- ^ Тейлор В. Р., Флорес Т. П., Оренго, Калифорния (1994). «Множественное выравнивание структуры белка». Белковая наука. 3 (10): 1858–70. Дои:10.1002 / pro.5560031025. ЧВК 2142613. PMID 7849601.
- ^ Оренго, Калифорния, Мичи А.Д., Джонс С., Джонс Д.Т., Суинделлс МБ, Торнтон Дж. М. (1997). «CATH: иерархическая классификация структур домена белка». Структура. 5 (8): 1093–1108. Дои:10.1016 / S0969-2126 (97) 00260-8. PMID 9309224.
- ^ а б Чжан И, Сколник Дж (2005). «TM-align: алгоритм выравнивания структуры белка на основе TM-score». Исследования нуклеиновых кислот. 33 (7): 2302–2309. Дои:10.1093 / нар / gki524. ЧВК 1084323. PMID 15849316.
- ^ Стефано Ангаран; Мэри Эллен Бок; Клаудио Гарутти; Конкеттина Герра1 (2009). «MolLoc: веб-инструмент для локального структурного выравнивания молекулярных поверхностей». Исследования нуклеиновых кислот. 37 (Выпуск веб-сервера): W565–70. Дои:10.1093 / нар / gkp405. ЧВК 2703929. PMID 19465382.
- ^ Гаэль Дебре; Арно Мартель; Филипп Куниасс (2009). «РАСМОТ-3D ПРО: поисковый веб-сервер по 3D-мотивам». Исследования нуклеиновых кислот. 37 (Выпуск веб-сервера): W459–64. Дои:10.1093 / нар / gkp304. ЧВК 2703991. PMID 19417073.
- ^ а б Александра Шульман-Пелег; Максим Шацкий; Рут Нусинов; Хаим Дж. Вольфсон (2008). «MultiBind и MAPPIS: веб-серверы для множественного выравнивания сайтов связывания белков 3D и их взаимодействия». Исследования нуклеиновых кислот. 36 (Web Server issue): W260–4. Дои:10.1093/nar/gkn185. ЧВК 2447750. PMID 18467424.
- ^ а б Alexandra Shulman-Peleg; Maxim Shatsky; Ruth Nussinov; Haim J Wolfson (2007). "Spatial chemical conservation of hot spot interactions in protein-protein complexes". BMC Биология. 5 (43): 43. Дои:10.1186/1741-7007-5-43. ЧВК 2231411. PMID 17925020.
- ^ Gabriele Ausiello; Pier Federico Gherardini; Paolo Marcatili; Anna Tramontano; Allegra Via; Manuela Helmer-Citterich (2008). "FunClust: a web server for the identification of structural motifs in a set of non-homologous protein structures". BMC Биология. 9: S2. Дои:10.1186/1471-2105-9-S2-S2. ЧВК 2323665. PMID 18387204.
- ^ Janez Konc; Dušanka Janežič (2010). "ProBiS algorithm for detection of structurally similar protein binding sites by local structural alignment". Биоинформатика. 26 (9): 1160–1168. Дои:10.1093/bioinformatics/btq100. ЧВК 2859123. PMID 20305268.
- ^ Hui Sun Lee; Wonpil Im (2012). "Identification of Ligand Templates using Local Structure Alignment for Structure-Based Drug Design". Journal of Chemical Information and Modeling. 52 (10): 2784–2795. Дои:10.1021/ci300178e. ЧВК 3478504. PMID 22978550.
- ^ Hui Sun Lee; Wonpil Im (2013). "Ligand Binding Site Detection by Local Structure Alignment and Its Performance Complementarity". Journal of Chemical Information and Modeling. 53 (9): 2462–2470. Дои:10.1021/ci4003602. ЧВК 3821077. PMID 23957286.
- ^ а б Barthel D., Hirst J.D., Blazewicz J., Burke E.K. and Krasnogor N. (2007). "ProCKSI: a decision support system for Protein (Structure) Comparison, Knowledge, Similarity and Information". BMC Bioinformatics. 8: 416. Дои:10.1186/1471-2105-8-416. ЧВК 2222653. PMID 17963510.CS1 maint: несколько имен: список авторов (связь)
- ^ Sippl, M.; Wiederstein, M. (2012). "Detection of spatial correlations in protein structures and molecular complexes". Структура. 20 (4): 718–728. Дои:10.1016/j.str.2012.01.024. ЧВК 3320710. PMID 22483118.
- ^ Torarinsson E, Sawera M, Havgaard JH, Fredholm M, Gorodkin J (2006). "Thousands of corresponding human and mouse genomic regions unalignable in primary sequence contain common RNA structure". Genome Res. 16 (7): 885–9. Дои:10.1101/gr.5226606. ЧВК 1484455. PMID 16751343.
- ^ Hoksza D, Svozil D (2012). "Efficient RNA pairwise structure comparison by SETTER method" (PDF). Биоинформатика. 28 (14): 1858–1864. Дои:10.1093/bioinformatics/bts301. PMID 22611129.
- ^ Cech P, Svozil D, Hoksza D (2012). "SETTER: web server for RNA structure comparison". Исследования нуклеиновых кислот. 40 (W1): W42–W48. Дои:10.1093/nar/gks560. ЧВК 3394248. PMID 22693209.
- ^ Havgaard JH, Lyngso RB, Stormo GD, Gorodkin J (2005). "Pairwise local structural alignment of RNA sequences with sequence similarity less than 40%". Биоинформатика. 21 (9): 1815–24. Дои:10.1093/bioinformatics/bti279. PMID 15657094.
- ^ Mathews DH, Turner DH (2006). "Prediction of RNA secondary structure by free energy minimization". Curr Opin Struct Biol. 16 (3): 270–8. Дои:10.1016/j.sbi.2006.05.010. PMID 16713706.
дальнейшее чтение
- Bourne PE, Shindyalov IN. (2003): Structure Comparison and Alignment. In: Bourne, P.E., Weissig, H. (Eds): Structural Bioinformatics. Hoboken NJ: Wiley-Liss. ISBN 0-471-20200-2
- Yuan X, Bystroff C. (2004) "Non-sequential Structure-based Alignments Reveal Topology-independent Core Packing Arrangements in Proteins", Биоинформатика. Nov 5, 2004
- Jung J, Lee B (2000). "Protein structure alignment using environmental profiles". Protein Eng. 13 (8): 535–543. Дои:10.1093/protein/13.8.535.
- Ye Y, Godzik A (2005). "Multiple flexible structure alignment using partial order graphs". Биоинформатика. 21 (10): 2362–2369. Дои:10.1093/bioinformatics/bti353. PMID 15746292.
- Sippl M, Wiederstein M (2008). "A note on difficult structure alignment problems". Биоинформатика. 24 (3): 426–427. Дои:10.1093/bioinformatics/btm622. PMID 18174182.