Болезнь накопления гликогена - Glycogen storage disease
| Болезнь накопления гликогена | |
|---|---|
| Другие имена | Гликогеноз, декстриноз |
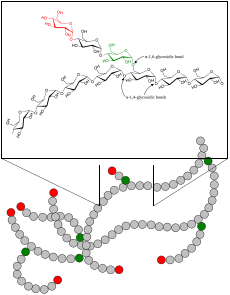 | |
| Гликоген | |
| Специальность | Эндокринология |
А болезнь накопления гликогена (GSD, также гликогеноз и декстриноз) это нарушение обмена веществ вызванный фермент недостатки затрагивая либо гликоген синтез, распад гликогена или гликолиз (расщепление глюкозы), обычно в мышцы и / или печень клетки.[1]
GSD имеет два класса причин: генетические и приобретенные. Генетическая GSD вызывается любым врожденная ошибка обмена веществ (генетически дефектный ферменты ) вовлечены в эти процессы. В животноводстве приобретенный GSD вызван: интоксикация с алкалоид кастаноспермин.[2]
Типы
| Тип (Эпоним) | Недостаточность ферментов (Ген[3]) | Заболеваемость (роды) | Гипо- гликемия ? | Гепато- мегалий ? | Hyperlip- идеемия ? | Мышечные симптомы | Развитие / прогноз | Другие симптомы |
|---|---|---|---|---|---|---|---|---|
| GSD 0 | Гликоген-синтаза (GYS2 ) | ? | да | Нет | Нет | Случайный мышечные спазмы | В некоторых случаях задержка роста | |
| GSD I / GSD 1 (болезнь фон Гирке ) | Глюкозо-6-фосфатаза (G6PC / SLC37A4 ) | 1 из 50 000–100 000[4][5] [6] | да | да | да | Никто | Нарушение роста | Лактоацидоз, гиперурикемия |
| GSD II / GSD 2 (Болезнь Помпе ) | Кислая альфа-глюкозидаза (GAA) | 1 из 13000. [7] | Нет | да | Нет | Мышечная слабость | Прогрессирующая слабость проксимальных скелетных мышц с различными временными рамками до порога функционального ограничения (от раннего детства до взрослого возраста). Примерно 15% популяции Помпе классифицируются как инфантильные Помпе, которые, как правило, смертельны в течение первого года при отсутствии лечения. | Сердечная недостаточность (младенческий), затрудненное дыхание (из-за мышечной слабости) |
| GSD III / GSD 3 (Болезнь Кори или Болезнь Форбса ) | Фермент разветвления гликогена (AGL ) | 1 из 100 000 | да | да | да | Миопатия | ||
| GSD IV / GSD 4 (Болезнь Андерсена ) | Фермент разветвления гликогена (GBE1 ) | 1 из 500 000[8] | Нет | Да, также цирроз | Нет | Миопатия и дилатационная кардиомиопатия | Неспособность процветать, смерть в возрасте ~ 5 лет | |
| GSD V / GSD 5 (Болезнь Макардла ) | Мышечная гликогенфосфорилаза (PYGM ) | 1 из 100 000–500 000[9][8] | Нет | Нет | Нет | Судороги, вызванные физической нагрузкой, Рабдомиолиз | Почечная недостаточность от миоглобинурия, явление второго ветра | |
| GSD VI / GSD 6 (Ее болезнь ) | Гликогенфосфорилаза печени (PYGL ) Мышечная фосфоглицератмутаза (PGAM2 ) | 1 из 65 000 - 85 000[10] | да | да | да [11] | Никто | первоначально доброкачественная, следует задержка развития. | |
| GSD VII / GSD 7 (Болезнь Таруи ) | Мышечная фосфофруктокиназа (ПФКМ ) | 1 из 1000000[12] | Нет | Нет | Нет | Мышечные спазмы и слабость, вызванные физической нагрузкой | отставание в развитии | В некоторых гемолитическая анемия |
| GSD IX / GSD 9 | Киназа фосфорилазы (PHKA2 / PHKB / PHKG2 / PHKA1 ) | ? | да | да | да | Никто | Задержка моторного развития, Отставание в развитии | |
| GSD X / GSD 10 | Фосфоглицерат мутаза (PGAM2 ) | ? | ? | ? | ? | Мышечные спазмы и слабость, вызванные физической нагрузкой | Миоглобинурия[13] | |
| GSD XI / GSD 11 | Лактатдегидрогеназа мышц (LDHA ) | ? | ? | ? | ? | |||
| Синдром Фанкони-Бикеля раньше GSD XI / GSD 11, больше не считается GSD | Транспортер глюкозы (GLUT2 ) | ? | да | да | Нет | Никто | ||
| GSD XII / GSD 12 (Дефицит альдолазы А ) | Альдолаза А (ALDOA ) | ? | Нет | В некоторых | Нет | Непереносимость физических упражнений, судороги. При некоторых рабдомиолизах. | Гемолитическая анемия и другие симптомы | |
| GSD XIII / GSD 13 | β-енолаза (ENO3 ) | ? | Нет | ? | Нет | Непереносимость физических упражнений, судороги | Увеличение интенсивности миалгии через десятилетия[14] | Сыворотка СК: Эпизодические возвышения; Снижается с отдыхом[14] |
| GSD XV / GSD 15 | Гликогенин-1 (GYG1 ) | Редко[15] | Нет | Нет | Нет | Атропия мышц | Медленно прогрессирующая слабость на протяжении десятилетий | Никто |
Примечания:
- Некоторые GSD имеют разные формы, например младенческий, ювенильный, взрослый (с поздним началом).
- Некоторые GSD имеют разные подтипы, например GSD1a / GSD1b, GSD9A1 / GSD9A2 / GSD9B / GSD9C / GSD9D.[3]
- GSD типа 0: Хотя гликогенсинтаза его дефицит не приводит к накоплению лишнего гликогена в печени, он часто классифицируется в GSD как тип 0, потому что это еще один дефект хранения гликогена, который может вызывать аналогичные проблемы.
- GSD типа VIII (GSD 8): в прошлом это считалось отдельным заболеванием,[16] однако теперь он классифицирован как GSD типа VI.[17] или GSD IXa1;[18] это было описано как Х-сцепленный рецессивный унаследовано.[19]
- GSD типа XI (GSD 11): Синдром Фанкони-Бикеля, гепаторенальный гликогеноз с почечным синдромом Фанкони, больше не считается болезнью накопления гликогена.[3]
- GSD типа XIV (GSD 14): теперь классифицируется как Врожденное нарушение гликозилирования тип 1 (CDG1T), влияет на фермент фосфоглюкомутазу (ген PGM1).[3]
- Болезнь Лафора считается сложным нейродегенеративным заболеванием, а также нарушением обмена гликогена.[20]
Диагностика
Эта секция нуждается в расширении. Вы можете помочь добавляя к этому. (Ноябрь 2017 г.) |
лечение
Лечение зависит от типа болезни накопления гликогена. GSD I обычно лечат частыми небольшими порциями углеводы и кукурузный крахмал, называется модифицированная терапия кукурузным крахмалом, чтобы предотвратить низкий уровень сахара в крови, в то время как другие методы лечения могут включать аллопуринол и колониестимулирующий фактор гранулоцитов человека.[21]
Эпидемиология
В целом, согласно исследованию в британская Колумбия примерно 2,3 ребенка на 100 000 рождений (1 из 43 000) страдают той или иной формой болезни накопления гликогена.[22] По оценкам, в Соединенных Штатах они встречаются у 1 на 20 000–25 000 рождений.[4] Уровень заболеваемости в Голландии оценивается в 1 на 40 000 новорожденных, в то время как в Мексике заболеваемость составляет 6,78 на 1000 новорожденных мужского пола.[23][24]
использованная литература
- ^ Cantú-Reyna, C .; Santos-Guzmán, J .; Cruz-Camino, H .; Vazquez Cantu, D.L .; Góngora-Cortéz, J.J .; Гутьеррес-Кастильо, А. (2019). «Заболеваемость дефицитом глюкозо-6-фосфатдегидрогеназы у латиноамериканского населения». Журнал неонатально-перинатальной медицины. 12 (2): 203–207. Дои:10.3233 / НПМ-1831. PMID 30741698.
- ^ Стегельмайер Б.Л., Молинье Р.Дж., Эльбейн А.Д., Джеймс Л.Ф. (май 1995 г.). «Поражения locoweed (Astragalus mollissimus), swainsonine и castanospermine у крыс». Ветеринарная патология. 32 (3): 289–98. Дои:10.1177/030098589503200311. PMID 7604496. S2CID 45016726.
- ^ а б c d Метаболизм гликогена themedicalbiochemistrypage.org
- ^ а б eMedicine Specialities> Заболевание накопления гликогена, тип I Автор: Карл С. Рот. Обновлено: 31 августа 2009 г.
- ^ The Association for Glycogen Storage Disease> Тип I Заболевание накопления гликогена Тип I GSD В архиве 2010-08-03 на Wayback Machine Октябрь 2006 г.
- ^ Cantú-Reyna, C .; Santos-Guzmán, J .; Cruz-Camino, H .; Vazquez Cantu, D.L .; Góngora-Cortéz, J.J .; Гутьеррес-Кастильо, А. (4 февраля 2019 г.). «Заболеваемость дефицитом глюкозо-6-фосфатдегидрогеназы у испаноязычного населения». Журнал неонатально-перинатальной медицины. 12 (2): 203–207. Дои:10.3233 / НПМ-1831. PMID 30741698.
- ^ https://pediatrics.aappublications.org/content/140/Supplement_1/S4
- ^ а б Стюарт, Грант; Ахмад, Наргис (2011). «Периоперационный уход за детьми с наследственными нарушениями обмена веществ». Повышение квалификации в области анестезии, неотложной помощи и боли. 11 (2): 62–68. Дои:10.1093 / bjaceaccp / mkq055.
- ^ http://mcardlesdisease.org/
- ^ eMedicine Specialities> Педиатрия: генетика и метаболические заболевания> Метаболические заболевания> Болезнь накопления гликогена типа VI Автор: Линн Иерарди-Курто, доктор медицинских наук. Обновлено: 4 августа 2008 г.
- ^ Гольдман, Ли; Шафер, Эндрю (2012). Лекарство Сесила Гольдмана (24-е изд.). Филадельфия: Эльзевьер / Сондерс. п. 1356. ISBN 978-1-4377-1604-7.
- ^ «База данных редких заболеваний». Orpha.net. Получено 2015-09-20.
- ^ Справка, Дом генетики. «Дефицит фосфоглицератмутазы». Домашний справочник по генетике. Получено 2019-02-06.
- ^ а б «Гликогенозы».
- ^ Малфатти Э., Нильссон Дж., Хедберг-Олдфорс С., Эрнандес-Лайн А., Мишель Ф., Домингес-Гонсалес С., Вьеннет Г., Акман Х.О., Корнблюм С., Ван ден Берг П., Ромеро Н. 2014) Новое заболевание накопления гликогена в мышцах, связанное с дефицитом гликогенина-1. Энн Нейрол 76 (6): 891-898
- ^ Людвиг М., Вольфсон С., Реннерт О. (октябрь 1972 г.). «Болезнь накопления гликогена, тип 8». Arch. Дис. Ребенок. 47 (255): 830–833. Дои:10.1136 / adc.47.255.830. ЧВК 1648209. PMID 4508182.
- ^ «Гликоген-накопительная болезнь типа VI: статья Линн Иерарди-Курто». EMedicine. 2019-02-02.
- ^ ЗАБОЛЕВАНИЕ ХРАНЕНИЯ ГЛИКОГЕНА IXa1; GSD9A1 OMIM - онлайн-менделевское наследование в человеке
- ^ «Определение: болезнь накопления гликогена типа VIII из медицинского онлайн-словаря».
- ^ Ortolano S, Vieitez I et al. Потеря корковых нейронов лежит в основе невропатологии болезни Лафора. Мол мозг 2014; 7: 7 ЧВК 3917365
- ^ «Заболевание накопления гликогена типа I - NORD (Национальная организация по редким заболеваниям)». NORD (Национальная организация по редким заболеваниям). Получено 23 марта 2017.
- ^ Эпплгарт Д.А., Тоун-младший, Лоури РБ (январь 2000 г.). «Частота врожденных нарушений метаболизма в Британской Колумбии, 1969–1996». Педиатрия. 105 (1): e10. Дои:10.1542 / пед.105.1.e10. PMID 10617747.
- ^ Cantú-Reyna, C .; Santos-Guzmán, J .; Cruz-Camino, H .; Vazquez Cantu, D.L .; Góngora-Cortéz, J.J .; Гутьеррес-Кастильо, А. (4 февраля 2019 г.). «Заболеваемость дефицитом глюкозо-6-фосфатдегидрогеназы у испаноязычного населения». Журнал неонатально-перинатальной медицины. 12 (2): 203–207. Дои:10.3233 / НПМ-1831. PMID 30741698.
- ^ Канту-Рейна, Консуэло; Зепеда, Луис Мануэль; Монтемайор, Рене; Бенавидес, Сантьяго; Гонсалес, Эктор Хавьер; Васкес-Канту, «Мерседес»; Крус-Камино, Эктор (27 сентября 2016 г.). «Частота врожденных нарушений обмена веществ при расширенном обследовании новорожденных в мексиканской больнице» (PDF). Журнал врожденных ошибок метаболизма и скрининга. 4: 232640981666902. Дои:10.1177/2326409816669027.
внешние ссылки
| Классификация |
|---|