Болезнь накопления гликогена III типа - Glycogen storage disease type III
| Болезнь накопления гликогена III типа | |
|---|---|
| Другие имена | Болезнь Кори, Дефицит Дебранчера, Болезнь Форбса, GSD III[1] |
 | |
| Микрофотография из болезнь накопления гликогена с гистологическими признаками, соответствующими болезни Кори. Биопсия печени. H&E пятно. | |
| Специальность | Эндокринология |
| Симптомы | Гипотония[2] |
| Причины | Мутация гена AGL[3] |
| Диагностический метод | Биопсия, Повышенные трансаминазы[4] |
| Уход | В настоящее время нет лекарства, Диетический режим[4] |
Болезнь накопления гликогена III типа является аутосомно-рецессивный нарушение обмена веществ и врожденная ошибка обмена веществ (в частности углеводы ) характеризуется дефицитом ферменты, разветвляющие гликоген.[3]
Он также известен как Болезнь Кори в честь нобелевских лауреатов 1947 г. Карл Кори и Герти Кори. Другие имена включают Болезнь Форбса в честь клинициста Гилберта Бернетта Форбса (1915–2003), американского врача, который далее описал особенности расстройства, или ограничить декстриноз, за счет предельных декстриноподобных структур в цитозоль.[2] Предел декстрин остается полимер, полученный после гидролиз гликогена. Без ферментов, разветвляющих гликоген, для дальнейшего превращения этих разветвленных полимеров гликогена в глюкозу, ограничивающий декстриноз аномально накапливается в цитоплазме.[5]
Гликоген - это молекула, которую организм использует для хранения углевод энергия. Симптомы GSD-III вызваны дефицитом фермента амило-1,6-глюкозидазы или фермент-дебранчер. Из-за этого избыточное количество аномального гликогена откладывается в печени, мышцах и, в некоторых случаях, в сердце.[требуется медицинская цитата ]
Признаки и симптомы
Болезнь накопления гликогена III типа проявляется во время младенчество с гипогликемия и неспособность процветать. Клиническое обследование обычно выявляет гепатомегалия. Мышечные заболевания, в том числе гипотония и кардиомиопатия, обычно происходит позже. Патология печени обычно регрессирует по мере того, как человек входит в юность, как и спленомегалия, если она у человека разовьется.[2]
Генетика
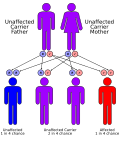
Что касается генетики, болезнь накопления гликогена III типа передается по наследству аутосомно-рецессивный (что означает, что оба родителя должны быть носителями) и встречается примерно у 1 из 100 000 живорождений. Самая высокая частота болезни накопления гликогена III типа наблюдается в Фарерские острова где это происходит в 1 из 3600 рождений, вероятно, из-за эффект основателя.[нужна цитата ] Кажется, есть две мутации в экзон 3 (c.17_18delAG) является одним из них, которые связаны с подтипом IIIb.[1][6]
В основе этого состояния лежат ген амило-альфа-1,6-глюкозидазы, 4-альфа-глюканотрансферазы и его мутации. Ген отвечает за создание фермент разветвления гликогена, что, в свою очередь, помогает в разложении гликогена.[3][7]
Диагностика
Что касается диагностики болезни накопления гликогена III типа, проводятся следующие тесты / экзамены, чтобы определить, есть ли у человека данное состояние:[8][9]
Дифференциальная диагностика
Дифференциальный диагноз болезни накопления гликогена III типа включает: GSD I, GSD IX и GSD VI. Однако это не означает, что нельзя различать и другие болезни накопления гликогена.[1]
Классификация
Клинические проявления болезни накопления гликогена III типа делятся на четыре класса:[3]
- GSD IIIa, является наиболее распространенным (наряду с GSD IIIb) и клинически включает мышца и печень участие
- GSD IIIb, который клинически имеет печень участие, но нет мышца участие
- GSD IIIc который клинически влияет на печень и мышцы.
- GSD IV влияет только на печень (не на мышцы)
Уход

Лечение болезни накопления гликогена типа III может включатьбелок диета, чтобы облегчить глюконеогенез. Дополнительно человеку могут понадобиться:[2][1][9]
- IV глюкоза (если пероральный путь не рекомендуется)
- Специалист по питанию
- Витамин Д (при остеопорозе / вторичном осложнении)
- Печеночный пересадка (при возникновении осложнений)
Рекомендации
- ^ а б c d Дагли, Адити; Sentner, Christiaan P .; Вайнштейн, Дэвид А. (1 января 1993 г.). «Болезнь накопления гликогена типа III». GeneReviews. PMID 20301788. Получено 11 августа 2016.обновление 2012
- ^ а б c d «Генетика болезни накопления гликогена III типа. Клиническая картина: история, физика, причины». emedicine.medscape.com. Получено 2016-08-11.
- ^ а б c d Справка, Дом генетики. «болезнь накопления гликогена III типа». Домашний справочник по генетике. Получено 2016-08-07.
- ^ а б «Болезнь накопления гликогена типа 3 | Информационный центр по генетическим и редким заболеваниям (GARD) - программа NCATS». rarediseases.info.nih.gov. Получено 2 января 2018.
- ^ Дж. Г. Салуэй (2012). Медицинская биохимия вкратце. Джон Вили и сыновья. п. 60. ISBN 9780470654514.
- ^ «Запись OMIM - № 232400 - Гликогеновая болезнь III; GSD3». www.omim.org. Получено 2016-08-11.
- ^ Справка, Дом генетики. «АГЛ». Домашний справочник по генетике. Получено 2016-08-11.
- ^ «Нарушения накопления гликогена. Врожденные нарушения обмена веществ | Пациент». Пациент. Получено 2016-08-11.
- ^ а б Кишнани, Priya S .; Остин, Стефани Л .; Арн, Памела; Бали, Дикша С .; Бони, Энн; Дело, Лаура Э .; Чанг, Венди К .; Desai, Dev M .; Эль-Гарбави, Ариг; Халлер, Рональд; Смит, Г. Питер А .; Смит, Аластер Д .; Hobson-Webb, Lisa D .; Векслер, Стефани Бернс; Вайнштейн, Дэвид А .; Уотсон, Майкл С. (1 июля 2010 г.). «Руководство по диагностике и лечению болезни накопления гликогена III типа». Генетика в медицине. 12 (7): 446–463. Дои:10.1097 / GIM.0b013e3181e655b6. ISSN 1098-3600. PMID 20631546.
дальнейшее чтение
- Майорандан, Себене; Мейер, Ута; Хартманн, Ганс; Дас, Аниб Мартин (1 января 2014 г.). «Болезнь накопления гликогена III типа: модифицированная диета Аткинса улучшает миопатию». Журнал редких заболеваний Orphanet. 9: 196. Дои:10.1186 / s13023-014-0196-3. ISSN 1750-1172. ЧВК 4302571. PMID 25431232.
- Sentner, Christiaan P .; Hoogeveen, Irene J .; Вайнштейн, Дэвид А .; Сантер, Рене; Мерфи, Элейн; Маккирнан, Патрик Дж .; Штойервальд, Ульрике; Beauchamp, Николас Дж .; Тайберт, Джоанна; Лафорет, Паскаль; Petit, François M .; Юбер, Орели; Лабрун, Филипп; Смит, Г. Питер А .; Деркс, Терри Дж. Дж. (22 апреля 2016 г.). «Болезнь накопления гликогена III типа: диагноз, генотип, лечение, клиническое течение и исход». Журнал наследственных метаболических заболеваний. 39 (5): 697–704. Дои:10.1007 / s10545-016-9932-2. ISSN 0141-8955. ЧВК 4987401. PMID 27106217.
внешняя ссылка
| Схолия имеет тема профиль для Болезнь накопления гликогена III типа. |
 СМИ, связанные с Болезнь накопления гликогена III типа в Wikimedia Commons
СМИ, связанные с Болезнь накопления гликогена III типа в Wikimedia Commons
| Классификация | |
|---|---|
| Внешние ресурсы |