Нуклеофильное ацильное замещение - Nucleophilic acyl substitution
Нуклеофильное ацильное замещение описать класс реакции замещения с привлечением нуклеофилы и ацил соединения. В этом типе реакции нуклеофил, такой как алкоголь, амин, или энолировать - вытесняет уходящая группа ацильного производного - такого как галогенид кислоты, ангидрид, или сложный эфир. Полученный продукт представляет собой карбонил -содержащее соединение, в котором нуклеофил занял место уходящей группы, присутствующей в исходном ацильном производном. Поскольку ацильные производные реагируют с широким спектром нуклеофилов и поскольку продукт может зависеть от конкретного типа ацильного производного и задействованного нуклеофила, реакции нуклеофильного ацильного замещения могут использоваться для синтеза множества различных продуктов.
Механизм реакции
Карбонильные соединения реагируют с нуклеофилами по механизму присоединения: нуклеофил атакует карбонильный углерод, образуя тетраэдрический промежуточный. Эту реакцию можно ускорить, если кислый условия, которые делают карбонил более электрофильный, или основной условия, которые обеспечивают более анионный и, следовательно, более реактивный нуклеофил. Сам тетраэдрический промежуточный продукт может представлять собой спирт или алкоксид, в зависимости от pH реакции.
Тетраэдрический интермедиат ацил соединение содержит заместитель прикрепленный к центральному углероду, который может действовать как уходящая группа. После образования тетраэдрического промежуточного соединения он разрушается, воссоздавая карбонильную связь C = O и выбрасывая уходящую группу в реакция элиминации. В результате этого двухстадийного процесса добавления / удаления нуклеофил занимает место уходящей группы в карбонильном соединении посредством промежуточного состояния, которое не содержит карбонил. Оба шага обратимый и в результате реакции нуклеофильного ацильного замещения являются равновесными процессами.[1][требуется полная цитата ] Поскольку равновесие будет благоприятствовать продукту, содержащему лучший нуклеофил, уходящая группа должна быть сравнительно бедным нуклеофилом, чтобы реакция была практичной.
Кислые условия
В кислых условиях карбонильная группа ацильного соединения 1 протонирован, что активирует его в направлении нуклеофильной атаки. На втором этапе протонированный карбонил 2 подвергается атаке нуклеофила (H-Z) с образованием тетраэдрического промежуточного соединения 3. Перенос протона от нуклеофила (Z) к уходящей группе (X) дает 4, который затем коллапсирует, выбрасывая протонированную уходящую группу (H-X), давая протонированное карбонильное соединение 5. Потеря протона дает продукт замещения, 6. Поскольку последняя стадия включает потерю протона, реакции нуклеофильного ацильного замещения считаются каталитическими в кислоте. Также обратите внимание, что в кислых условиях нуклеофил обычно будет существовать в протонированной форме (то есть H-Z вместо Z−).

Основные условия
Под основной условиях, нуклеофил (Nuc) атакует карбонильную группу ацильного соединения 1 с получением тетраэдрического промежуточного алкоксида 2. Промежуточный продукт коллапсирует и вытесняет уходящую группу (X) с образованием продукта замещения 3. Хотя реакции нуклеофильного ацильного замещения могут катализироваться основанием, реакция не произойдет, если уходящая группа является более сильным основанием, чем нуклеофил (т.е. уходящая группа должна иметь более высокое значение pKа чем нуклеофил). В отличие от процессов, катализируемых кислотой, как нуклеофил, так и уходящая группа существуют в виде анионов в основных условиях.

Этот механизм поддерживается маркировка изотопов эксперименты. Когда этилпропионат с кислород-18 -меченная этокси-группа обрабатывается гидроксид натрия (NaOH) метка кислород-18 полностью отсутствует в пропионовая кислота и встречается исключительно в этиловый спирт.[2]

Тенденции реактивности
Существует пять основных типов ацильных производных. Галогениды кислот являются наиболее реактивными по отношению к нуклеофилам, за ними следуют ангидриды, сложные эфиры, и амиды. Карбоксилат ионы практически не реагируют с нуклеофильным замещением, так как не имеют уходящей группы. Реакционная способность этих пяти классов соединений охватывает широкий диапазон; относительные скорости реакции хлорангидридов и амидов различаются в 10 раз.13.[3]

Основным фактором в определении реакционной способности ацильных производных является способность уходящей группы, которая связана с кислотностью. Слабые базы лучше уходят из группы, чем сильные; вид с сильным конъюгированная кислота (например. соляная кислота ) будет лучше уходящей группой, чем виды со слабой конъюгированной кислотой (например, уксусная кислота ). Таким образом, хлористый ion - лучшая уходящая группа, чем ацетат-ион. Реакционная способность ацильных соединений по отношению к нуклеофилам снижается с увеличением основности уходящей группы, как показано в таблице.[4]
| Название соединения | Структура | Выход из группы | пKа конъюгированной кислоты |
|---|---|---|---|
| Ацетилхлорид |  | −7 | |
| Уксусный ангидрид | 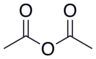 |  | 4.76 |
| Этилацетат |  | 15.9 | |
| Ацетамид |  | 38 | |
| Ацетат анион | | Нет данных | Нет данных |

Еще одним фактором, который играет роль в определении реакционной способности ацильных соединений, является резонанс. Амиды проявляют две основные резонансные формы. Оба они вносят основной вклад в общую структуру, так что амидная связь между карбонильным углеродом и амидным азотом имеет существенное значение. двойная связь характер. Энергетический барьер для вращения вокруг амидной связи составляет 75–85 кДж / моль (18–20 ккал / моль), что намного больше, чем значения, наблюдаемые для обычных одинарных связей. Например, связь C – C в этане имеет энергетический барьер всего 12 кДж / моль (3 ккал / моль).[3] Как только нуклеофил атакует и образуется тетраэдрический промежуточный продукт, энергетически выгодный резонансный эффект теряется. Это помогает объяснить, почему амиды являются одними из наименее реакционноспособных ацильных производных.[4]
Сложные эфиры демонстрируют меньшую стабилизацию резонанса, чем амиды, поэтому образование тетраэдрического промежуточного соединения и последующая потеря резонанса не так энергетически невыгодны. Ангидриды испытывают даже более слабую стабилизацию резонанса, поскольку резонанс разделен между двумя карбонильными группами, и они более реакционноспособны, чем сложные эфиры и амиды. В галогенангидридах очень мало резонанса, поэтому энергетический штраф за образование тетраэдрического промежуточного соединения невелик. Это помогает объяснить, почему галогенангидриды являются наиболее реакционноспособными производными ацила.[4]
Реакции ацильных производных
Многие реакции нуклеофильного ацильного замещения включают превращение одного ацильного производного в другое. В общем, для практического применения преобразования между ацильными производными должны происходить от относительно реакционноспособного соединения до менее реакционноспособного; хлорангидрид можно легко превратить в сложный эфир, но преобразование сложного эфира непосредственно в хлорангидрид практически невозможно. При преобразовании между ацильными производными продукт всегда будет более стабильным, чем исходное соединение.
Также возможны реакции нуклеофильного ацильного замещения, которые не включают взаимное превращение между ацильными производными. Например, амиды и карбоновые кислоты реагируют с Реактивы Гриньяра для производства кетонов. Здесь представлен обзор реакций, в которых может участвовать каждый тип ацильного производного.
Галогениды кислот
Галогениды кислот являются наиболее реакционноспособными производными ацила и могут легко превращаться в любые другие. Галогенангидриды реагируют с карбоновыми кислотами с образованием ангидридов. Если структура кислоты и хлорангидрида различается, продукт представляет собой смешанный ангидрид. Во-первых, карбоновая кислота атакует хлорангидрид (1) с образованием тетраэдрического промежуточного 2. Тетраэдрический промежуточный продукт коллапсирует, выбрасывая хлорид-ион в качестве уходящей группы и образуя оксоний виды 3. Депротонирование дает смешанный ангидрид, 4, и эквивалент HCl.

Спирты и амины реагировать с галогенангидридами с образованием сложные эфиры и амиды соответственно, в реакции, формально известной как Реакция Шоттена-Баумана.[5] Галогенангидриды гидролизуются в присутствии воды с образованием карбоновых кислот, но этот тип реакции редко бывает полезен, поскольку карбоновые кислоты обычно используются для синтеза галогенангидридов. Большинство реакций с галогенангидридами проводят в присутствии ненуклеофильного основания, такого как пиридин, чтобы нейтрализовать галогеноводородную кислоту, образующуюся в качестве побочного продукта.
Галогениды кислоты будут реагировать с нуклеофилами углерода, такими как Гриньярс и енолирует, хотя может возникнуть смесь продуктов. Хотя углеродный нуклеофил сначала реагирует с галогенангидридом с образованием кетона, кетон также подвержен нуклеофильной атаке и может быть преобразован в третичный спирт. Например, когда бензоилхлорид (1) обрабатывают двумя эквивалентами реактива Гриньяра, такими как метилмагнийбромид (MeMgBr), 2-фенил-2-пропанол (3) получается с отличным урожаем. Несмотря на то что ацетофенон (2) является промежуточным продуктом в этой реакции, его невозможно изолировать, поскольку он быстро реагирует со вторым эквивалентом MeMgBr после образования.[6]


В отличие от большинства других углеродных нуклеофилов, диалкилкупраты лития, часто называемые Реактивы Гилмана - можно добавить к галогенангидридам только один раз, чтобы получить кетоны. Однако реакция между галогенангидридом и реагентом Гилмана не является реакцией нуклеофильного ацильного замещения и, как полагают, протекает по радикальному пути.[2] В Синтез кетона Вайнреба также можно использовать для преобразования галогенангидридов в кетоны. В этой реакции галогенангидрид сначала превращается в N-метокси-N-метиламид, известный как амид Вайнреба. Когда углеродный нуклеофил - например, Гриньяр или литийорганический реагент - добавляет к амиду Вайнреба, металл хелатный карбонильными и N-метокси-атомами кислорода, предотвращая дальнейшие нуклеофильные присоединения.[7]
в Ацилирование Фриделя – Крафтса галогенангидриды действуют как электрофилы для электрофильное ароматическое замещение. А Кислота Льюиса - такие как хлорид цинка (ZnCl2), хлорид железа (III) (FeCl3), или хлорид алюминия (AlCl3) - координируется с галогеном на галогенангидриде, активируя соединение в направлении нуклеофильной атаки со стороны активирован ароматическое кольцо. Для ароматических колец, особенно богатых электронами, реакция протекает без кислоты Льюиса.[8]
Тиоэфиры
Химия тиоэфиры и галогенангидриды аналогичны, реактивность Напоминает, но мягче, чем хлорангидриды.
Ангидриды
Химический состав галогенангидридов и ангидридов аналогичен. Хотя ангидриды не могут быть преобразованы в галогенангидриды, они могут быть преобразованы в оставшиеся ацильные производные. Ангидриды также участвуют в реакциях типа Шоттена-Баумана для получения сложных эфиров и амидов из спиртов и аминов, а вода может гидролизовать ангидриды до их соответствующих кислот. Как и в случае галогенангидридов, ангидриды могут также реагировать с нуклеофилами углерода с образованием кетонов и / или третичных спиртов и могут участвовать как в ацилировании Фриделя-Крафтса, так и в синтезе кетонов Вайнреба.[8] Однако, в отличие от галогенангидридов, ангидриды не реагируют с реагентами Гилмана.[2]
Реакционную способность ангидридов можно увеличить, используя каталитическое количество N, N-диметиламинопиридин, или DMAP. Пиридин также может использоваться для этой цели и действует через аналогичный механизм.[5]

Во-первых, DMAP (2) атакует ангидрид (1) с образованием тетраэдрического интермедиата, который коллапсирует, чтобы удалить карбоксилат-ион с образованием амида 3. Этот промежуточный амид более активен в отношении нуклеофильной атаки, чем исходный ангидрид, потому что диметиламинопиридин является более удаляемой группой, чем карбоксилат. В последнем наборе шагов нуклеофил (Nuc) атакует 3 дать еще одно тетраэдрическое промежуточное соединение. Когда этот промежуточный продукт разрушается, давая продукту 4пиридиновая группа удаляется и ее ароматичность восстанавливается - мощная движущая сила и причина, по которой соединение пиридина является более удаляемой группой, чем ион карбоксилата.
Сложные эфиры
Сложные эфиры менее реакционноспособны, чем галогенангидриды и ангидриды. Как и более реакционноспособные ацильные производные, они могут реагировать с аммиак и первичные и вторичные амины для получения амидов, хотя этот тип реакции не часто используется, поскольку галогенангидриды дают лучшие выходы. Сложные эфиры можно превратить в другие сложные эфиры в процессе, известном как переэтерификация. Переэтерификация может быть катализируемой кислотой или основанием и включает реакцию сложного эфира со спиртом. К сожалению, поскольку уходящая группа также является спиртом, прямые и обратные реакции часто будут происходить с одинаковой скоростью. Используя большой избыток реагент спирт или удаление алкоголя из уходящей группы (например, через дистилляция ) будет способствовать продвижению реакции к завершению в соответствии с Принцип Ле Шателье.[9]
Катализируемый кислотой гидролиз сложных эфиров также является равновесным процессом - по сути, обратным Этерификация Фишера реакция. Поскольку спирт (который действует как уходящая группа) и вода (которая действует как нуклеофил) имеют одинаковые значения pKа значения, прямая и обратная реакции конкурируют друг с другом. Как и в случае переэтерификации, использование большого избытка реагента (воды) или удаление одного из продуктов (спирта) может способствовать прямой реакции.
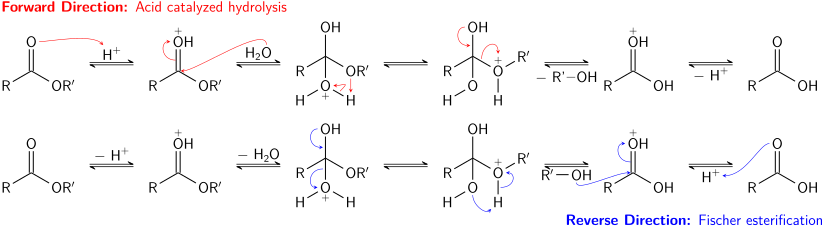
Основной гидролиз сложных эфиров, известный как омыление, не является равновесным процессом; в реакции расходуется полный эквивалент основания, в результате чего образуется один эквивалент спирта и один эквивалент карбоксилатной соли. Омыление сложных эфиров жирные кислоты это промышленно важный процесс, используемый при производстве мыла.[9]
Сложные эфиры могут вступать в различные реакции с нуклеофилами углерода. Как и в случае галогенангидридов и ангидридов, они будут реагировать с избытком реактива Гриньяра с образованием третичных спиртов. Сложные эфиры также легко реагируют с енолирует. в Клейзеновская конденсация, енолят одного сложного эфира (1) будет атаковать карбонильную группу другого сложного эфира (2) с образованием тетраэдрического промежуточного 3. Промежуточный продукт разрушается, вытесняя алкоксид (R'O−) и получения β-кетоэфира 4.

Возможны также перекрестные конденсации Клайзена, в которых енолят и нуклеофил являются разными сложными эфирами. An внутримолекулярный Клейзеновская конденсация называется Конденсация Дикмана или циклизация Дикмана, поскольку ее можно использовать для образования колец. Сложные эфиры также могут конденсироваться с енолями кетонов и альдегидов с образованием β-дикарбонильных соединений.[10] Конкретным примером этого является Перегруппировка Бейкера – Венкатарамана, в котором ароматический орто-ацилоксикетон подвергается внутримолекулярному нуклеофильному ацильному замещению и последующей перегруппировке с образованием ароматического β-дикетона.[11] В Перестановка Чана представляет собой другой пример перегруппировки, являющейся результатом реакции внутримолекулярного нуклеофильного ацильного замещения.
Амиды
Из-за их низкой реактивности амиды не участвуют почти в таком количестве реакций нуклеофильного замещения, как другие ацильные производные. Амиды устойчивы к воде и примерно в 100 раз более устойчивы к гидролиз чем сложные эфиры.[3] Однако амиды можно гидролизовать до карбоновых кислот в присутствии кислоты или основания. Стабильность амидные связи имеет биологические последствия, поскольку аминокислоты которые составляют белки связаны амидными связями. Амидные связи достаточно устойчивы к гидролизу, чтобы поддерживать структуру белка в водный окружающей среды, но достаточно восприимчивы, чтобы их можно было сломать при необходимости.[3]
Первичные и вторичные амиды неблагоприятно реагируют с нуклеофилами углерода. Реактивы Гриньяра а литийорганические соединения будут действовать как основания, а не нуклеофилы, и будут просто депротонировать амид. Третичные амиды не испытывают этой проблемы и реагируют с нуклеофилами углерода с образованием кетоны; то амид анион (NR2−) является очень сильным основанием и, следовательно, очень плохой уходящей группой, поэтому нуклеофильная атака происходит только один раз. При реакции с углеродными нуклеофилами N,N-диметилформамид (DMF) можно использовать для введения формил группа.[12]

Вот, фениллитий 1 атакует карбонильную группу ДМФ 2, давая тетраэдрический промежуточный 3. Поскольку диметиламидный анион является плохо уходящей группой, промежуточное соединение не разрушается, и другое нуклеофильное присоединение не происходит. При кислотной обработке алкоксид протонируется с образованием 4, затем амин протонируется с образованием 5. Удаление нейтральной молекулы диметиламин и потеря протона дает бензальдегид, 6.
Карбоновые кислоты
Карбоновые кислоты не особенно реактивны по отношению к нуклеофильному замещению, хотя они могут быть превращены в другие ацильные производные. Превращение карбоновой кислоты в амид возможно, но не так просто. Вместо того чтобы действовать как нуклеофил, амин будет реагировать как основание в присутствии карбоновой кислоты с образованием аммония. карбоксилат поваренная соль. Нагревание соли до температуры выше 100 ° C приведет к удалению воды и образованию амида. Этот метод синтеза амидов имеет промышленное значение, а также имеет лабораторное применение.[13] В присутствии сильного кислотного катализатора карбоновые кислоты могут конденсировать с образованием ангидридов кислот. Однако в результате конденсации образуется вода, которая может гидролизовать ангидрид до исходных карбоновых кислот. Таким образом, образование ангидрида посредством конденсации является равновесным процессом.
В условиях кислотного катализатора карбоновые кислоты будут реагировать со спиртами с образованием сложные эфиры через Этерификация Фишера реакция, которая также является равновесным процессом. В качестве альтернативы, диазометан может использоваться для превращения кислоты в сложный эфир. Хотя реакции этерификации с диазометаном часто дают количественные выходы, диазометан пригоден только для образования метиловых эфиров.[13]
Тионил хлорид могут быть использованы для превращения карбоновых кислот в их соответствующие ацилхлориды. Во-первых, карбоновая кислота 1 атакует тионилхлорид, а уходит хлорид-ион. Результирующий оксониевый ион 2 активируется в направлении нуклеофильной атаки и имеет хорошую уходящую группу, что отличает ее от нормальной карбоновой кислоты. На следующем этапе 2 подвергается атаке хлорид-ионом с образованием тетраэдрического промежуточного соединения 3, хлорсульфит. Тетраэдрический интермедиат схлопывается с потерей диоксид серы и хлорид-ион, давая протонированный ацилхлорид 4. Хлорид-ион может удалить протон в карбонильной группе, давая ацилхлорид 5 с потерей HCl.

Хлорид фосфора (III) (PCl3) и хлорид фосфора (V) (PCl5) также будет преобразовывать карбоновые кислоты в хлорангидриды по аналогичному механизму. Один эквивалент PCl3 может реагировать с тремя эквивалентами кислоты, образуя один эквивалент H3PO3, или фосфорная кислота, в дополнение к желаемому хлорангидриду. PCl5 реагирует с карбоновыми кислотами в соотношении 1: 1 и производит оксихлорид фосфора (V) (POCl3) и хлористый водород (HCl) в качестве побочных продуктов.
Карбоновые кислоты реагируют с реактивами Гриньяра и литийорганическими соединениями с образованием кетонов. Первый эквивалент нуклеофила действует как основание и депротонирует кислоту. Второй эквивалент атакует карбонильную группу, создавая близнец алкоксид дианиона, который при обработке протонируется с образованием гидрата кетона. Поскольку большинство кетоновых гидратов нестабильны по сравнению с соответствующими кетонами, равновесие между ними сильно смещено в пользу кетона. Например, константа равновесия образования ацетон гидрат из ацетона составляет всего 0,002. Карбоксильная группа является наиболее кислой в органических соединениях.[14]
Смотрите также
использованная литература
- ^ Уэйд, 2010, стр. 996–997.
- ^ а б c Макмерри, Джон (1996). Органическая химия (4-е изд.). Пасифик Гроув, Калифорния: Издательство Brooks / Cole Publishing Company. стр.820–821. ISBN 0534238327.
- ^ а б c d Кэри, Фрэнсис А. (2006). Органическая химия (6-е изд.). Нью-Йорк: Макгроу-Хилл. стр.866–868. ISBN 0072828374.
- ^ а б c Уэйд, 2010, стр. 998–999.
- ^ а б Курти, Ласло; Барбара Чако (2005). Стратегические применения названных реакций в органическом синтезе. Лондон: Elsevier Academic Press. п. 398. ISBN 0124297854.
- ^ Макмерри, 1996, стр. 826–827.
- ^ Курти и Чако 2005, стр. 478.
- ^ а б Курти и Чако 2005, стр. 176.
- ^ а б Уэйд, 2010, стр. 1005–1009.
- ^ Кэри 2006, стр. 919–924.
- ^ Курти и Чако 2005, стр. 30.
- ^ Алан Р. Катрицки; Мет-Кон, Отто; Чарльз Рис, ред. (1995). Комплексные преобразования органических функциональных групп. 3 (1-е изд.). Оксфорд: Pergamon Press. п.90. ISBN 0080423248.
- ^ а б Уэйд, 2010, стр. 964–965.
- ^ Уэйд 2010, стр. 838.
внешние ссылки
- Реакция уксусный ангидрид с участием ацетон в Органический синтез Coll. Vol. 3, стр. 16; Vol. 20, стр. 6 Статья