Диоксигеназа - Dioxygenase
| Диоксигеназа | |||||||||
|---|---|---|---|---|---|---|---|---|---|
 кристаллическая структура acinetobacter sp. adp1 протокатехуат 3,4-диоксигеназа в комплексе с 3,4-дигидроксибензоатом | |||||||||
| Идентификаторы | |||||||||
| Символ | Диоксигеназа_C | ||||||||
| Pfam | PF00775 | ||||||||
| Pfam клан | CL0287 | ||||||||
| ИнтерПро | IPR000627 | ||||||||
| PROSITE | PDOC00079 | ||||||||
| SCOP2 | 2шт / Объем / СУПФАМ | ||||||||
| |||||||||
Диоксигеназы находятся оксидоредуктаза ферменты. Аэробная жизнь, из простых одноклеточных бактерии виды в комплекс эукариотический организмов, эволюционировал, чтобы зависеть от окислительной способности дикислород в различных метаболических путях. От энергичного аденозинтрифосфат (ATP) генерация ксенобиотик деградации, использование двуокиси кислорода в качестве биологического окислитель широко распространена и разнообразна по точному механизму ее использования. Ферменты используют множество различных схем использования дикислорода, и это во многом зависит от субстрат и реакция под рукой.
Сравнение с монооксигеназами
в монооксигеназы, только один атом кислорода включается в субстрат, а другой восстанавливается до молекулы воды. Диоксигеназы (EC 1.13.11 ) катализируют окисление субстрата без восстановления одного атома кислорода из двуокиси кислорода до молекулы воды. Однако это определение неоднозначно, поскольку оно не принимает во внимание, сколько субстратов участвует в реакции. Большинство диоксигеназ полностью объединяют дикислород в один субстрат, и множество кофактор схемы используются для достижения этого. Например, в α-кетоглутарат -зависимые ферменты, один атом дикислорода включен в два субстрата, один из которых всегда является α-кетоглутаратом, и эта реакция вызывается одноядерным железным центром.
Железосодержащие ферменты
Наиболее широко наблюдаемый кофактор, участвующий в реакциях диоксигенации, - это утюг, но каталитический Схема, используемая этими железосодержащими ферментами, весьма разнообразна. Железосодержащие диоксигеназы можно подразделить на три класса в зависимости от того, как железо включается в активный центр: те, которые используют моноядерный центр железа, те, которые содержат Rieske [2Fe-2S], и те, которые используют гем протезная группа.
Мононуклеарные диоксигеназы железа
Одноядерные диоксигеназы железа или неядерныегем железозависимые диоксигеназы, как их еще называют, все используют одно каталитическое железо для включения одного или обоих атомов кислорода в субстрат. Несмотря на это обычное явление оксигенации, одноядерные диоксигеназы железа различаются по способу использования активации диоксигена для ускорения определенных химических реакций.[1] Например, расщепление углерод-углеродной связи, гидропероксидирование жирных кислот, разрыв углерод-серной связи и окисление тиола - все это реакции, катализируемые моноядерными диоксигеназами железа.[1][2][3]
Большинство одноядерных диоксигеназ железа входят в состав надсемейство купинов в котором общая доменная структура описывается как шестицепочечная β-бочковая складка (или желе-ролл мотив). В центре этой бочкообразной структуры находится ион металла, чаще всего двухвалентного железа, координационное окружение которого часто обеспечивается остатками в двух частично консервативных структурных мотивах: G (X)5HXH (X)3-4БЫВШИЙ)6G и G (X)5-7PXG (X)2H (X)3Н.[4][5]


Двумя важными группами мононуклеарных негемовых диоксигеназ железа являются катехолдиоксигеназы и 2-оксоглутарат (2OG) -зависимые диоксигеназы.[6] В катехолдиоксигеназы, некоторые из наиболее хорошо изученных ферментов диоксигеназы, используют дикислород для расщепления углерод-углеродной связи ароматической катехол кольцевая система.[4] Катехолдиоксигеназы далее классифицируются как «экстрадиол» или «интрадиол», и это различие основано на механистических различиях в реакциях (рисунки 1 и 2). Ферменты внутрирадиола расщепляют углерод-углеродную связь между двумя гидроксильными группами. Активный центр трехвалентного железа координируется четырьмя белковыми лигандами - двумя. гистидин и два остатки тирозината - тригонально-бипирамидным образом с молекулой воды в пятом координационном центре.[3] Как только катехолатный субстрат связывается с металлическим центром в двузубый За счет депротонированных гидроксильных групп трехвалентное железо «активирует» субстрат посредством отвода электрона с образованием радикальный на подложке. Затем это позволяет протекать реакции с кислородом и последующему расщеплению внутри диола через промежуточный циклический ангидрид.[2][4] Члены экстрадиола используют двухвалентное железо в качестве активного окислительно-восстановительного состояния, и этот центр обычно координируется октаэдрически через мотив 2-His-1-Glu с лабильными водными лигандами, занимающими пустые позиции. Как только субстрат связывается с центром железа, это способствует связыванию дикислорода и последующей активации.[2][4][7] Затем эта активированная форма кислорода вступает в реакцию с субстратом, в конечном итоге разрывая связь углерод-углерод, прилегающую к гидроксильным группам, посредством образования промежуточного α-кетолактона.[3]
В 2OG-зависимых диоксигеназах двухвалентное железо (Fe (II) ) также координируется мотивом «лицевой триады» (His) 2 (Glu / Asp) 1. Бидентатная координация 2OG и воды завершает псевдооктаэдрическую координационную сферу. После связывания субстрата водный лиганд высвобождается, образуя открытый координационный сайт для активации кислорода.[6] При связывании кислорода происходит малоизученная трансформация, во время которой 2OG окислительно декарбоксилируется до сукцината, и связь O-O расщепляется с образованием Fe (IV) -oxo (Феррил ) средний. Этот мощный окислитель затем используется для проведения различных реакций, включая гидроксилирование, галогенирование и деметилирование.[8] В наиболее охарактеризованном случае гидроксилазы, промежуточное соединение феррила, отрывают атом водорода от целевого положения субстрата, образуя радикал субстрата и Fe (III) -OH. Затем этот радикал соединяется с гидроксидным лигандом, образуя гидроксилированный продукт и состояние покоя фермента Fe (II).[8]
Диоксигеназы Риске
Диоксигеназы Риске катализируют цис-дигидроксилирование аренов до продуктов цис-дигидродиола. Эти ферменты широко присутствуют в почвенных бактериях, таких как Псевдомонады,[3] и их реакции составляют начальную стадию биоразложения ароматических углеводородов.[2] Диоксигеназы Риске имеют более сложную структуру, чем другие диоксигеназы, из-за необходимости эффективного пути переноса электронов (рис. 2), обеспечивающего дополнительное одновременное двухэлектронное восстановление ароматического субстрата.

Каталитически компетентная диоксигеназа Риске состоит из трех компонентов: НАДН-зависимая ФАД редуктаза, а ферредоксин с двумя кластерами [2Fe-2S] Риске и α3β3 оксигеназой, каждая α-субъединица которой содержит одноядерный центр железа и кластер [2Fe-2S] Риске.[2] Внутри каждой α-субъединицы кластер железо-сера и одноядерный центр железа разделены расстоянием примерно в 43 Å, что слишком далеко для эффективной работы. перенос электронов происходить. Вместо этого предполагается, что перенос электронов осуществляется через эти два центра в соседних субъединицах, что железо-серный кластер одной субъединицы передает электроны моноядерному железному центру соседней субъединицы, который удобно разделен на ~ 12 Å. Хотя это расстояние кажется оптимальным для эффективного переноса электронов, замена мостикового остатка аспартата вызывает потерю функции фермента, предполагая, что перенос электронов вместо этого происходит через сеть водородных связей, удерживаемую этим остатком аспартата.[3]

Механистическая картина для этого класса диоксигеназ еще не ясна, но есть доказательства, подтверждающие наличие промежуточного соединения гидроперокси железа (III) в пути реакции.[7] Эта разновидность может представлять активный окислитель или может подвергаться гемолитическому разрыву связи O-O с образованием промежуточного соединения железа (V) -оксо в качестве рабочего окислителя.[3][7] Диоксигеназа Риске представляет собой мощный класс окислительно-восстановительных ферментов, и в дополнение к диоксигенации сообщалось о таких реакциях, как сульфоксидирование, десатурация и бензильное окисление.[2]
Гемсодержащие диоксигеназы
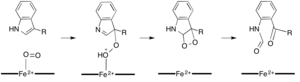
В то время как большинство железозависимых диоксигеназ используют негемовый кофактор железа, окисление L- (и D-) триптофана до N-формилкинуренина катализируется либо триптофан-2,3-диоксигеназа (TDO) или индоламин-2,3-диоксигеназа (IDO), которые представляют собой гем-диоксигеназы, которые используют железо, координируемое простетической группой гема B.[9][10] Хотя эти диоксигеназы представляют интерес отчасти потому, что они уникально используют гем для катализа, они также представляют интерес из-за их важности в триптофан регуляция в клетке, что имеет множество физиологических последствий.[11] Считается, что начальная ассоциация субстрата с диоксидом железа в активном центре фермента происходит либо посредством радикального, либо электрофильного присоединения, для чего требуется двухвалентное или трехвалентное железо соответственно.[9] Хотя точный механизм реакции гем-зависимых диоксигеназ все еще обсуждается, предполагается, что реакция протекает через диоксетан или Криджи механизм (рисунки 4, 5).[9][11]

Камбиалистические диоксигеназы
Хотя железо является наиболее распространенным кофактором, используемым для ферментативной диоксигенации, оно не требуется для всех диоксигеназ для катализа. Кверцетин 2,3-диоксигеназа (кверцетиназа, QueD) катализирует диоксигенолитическое расщепление кверцетин к 2-протокатехуоилфлороглюцинолкарбоновой кислоте и монооксид углерода.[12] Наиболее охарактеризованный фермент из Аспергиллы japonicus, требует наличия медь,[4] и были обнаружены бактериальные кверцетиназы, которые имеют довольно беспорядочный (камбиалистический)[13] в их требованиях к металлическому центру, с различной степенью активности сообщается с заменой двухвалентный марганец, кобальт, утюг, никель и медь.[12] (Кверцетин, роль в обмене веществ).Ациредуктон (1,2-дигидрокси-5- (метилтио) пент-1-ен-3-он) диоксигеназа (ARD) встречается в обоих прокариоты и эукариоты.[4][12][14] Ферменты ARD из большинства видов связывают двухвалентное железо и катализируют окисление ациредуктона до 4- (метилтио) -2-оксобутаноата, α-кетокислоты метионин, и муравьиная кислота. Тем не мение, ARD из Клебсиелла окситока катализирует дополнительную реакцию, когда никель (II) связан: вместо этого он производит 3- (метилтио) пропионат, формиат и монооксид углерода в результате реакции ациредуктона с кислородом. Активность Fe-ARD тесно переплетается с механизмом спасения метионина, в котором метилтиоаденозиновый продукт клеточного S-аденозил метионин (SAM) реакции в конечном итоге превращаются в ациредуктон.
Хотя точная роль Ni-ARD неизвестна, предполагается, что он помогает регулировать уровень метионина, действуя как шунт в пути спасения. Этот К. окситока Фермент представляет собой уникальный пример, когда присутствующий ион металла определяет, какая реакция катализируется. Кверцетиназы и ферменты ARD являются членами надсемейство купинов, к которым также относятся одноядерные ферменты железа.[15] Схема координации металлов для ферментов QueD представляет собой либо 3-His, либо 3-His-1-Glu, при этом точное расположение зависит от организма.[4] Все ферменты ARD хелат каталитический металл (Ni или Fe) через мотив 3-His-1-Glu.[15] В этих диоксигеназах координирующая лиганды представлены обоими типичными купиновыми мотивами. В ферментах ARD металл существует в октаэдрическое расположение с тремя гистидин остатки, составляющие лицевую триаду.[14] Металлические центры бактериальной кверцетиназы обычно имеют тригонально-бипирамидный или октаэдрическая координационная среда, когда имеется четыре белковых лиганда; Металлические центры медьзависимых ферментов QueD обладают искаженной тетраэдрической геометрией, в которой только три консервативных остатка гистидина обеспечивают координационные лиганды.[4][12] Пустые координационные центры во всех металлических центрах заняты аквалигандами до тех пор, пока они не будут замещены поступающим субстратом.
Способность этих диоксигеназ сохранять активность в присутствии кофакторов других металлов с широким диапазоном значений редокс потенциалов предполагает, что металлический центр не играет активной роли в активации двуокиси кислорода. Скорее, считается, что металлический центр удерживает субстрат в правильной геометрии, чтобы он мог реагировать с кислородом. В этом отношении эти ферменты напоминают интрадиол. катехолдиоксигеназы при этом металлические центры активируют субстрат для последующей реакции с кислородом.
Кофакторнезависимые диоксигеназы

Диоксигеназы, катализирующие реакции без необходимости в кофакторе, встречаются в природе гораздо реже, чем те, которые действительно нуждаются в них. Две диоксигеназы, 1H-3-гидрокси-4-оксохинолин 2,4-диоксигеназа (QDO) и 1H-3-гидрокси-4-оксохинальдин 2,4-диоксигеназа (HDO), как было показано, не требует ни органического, ни металлического кофактора.[16] Эти ферменты катализируют деградацию хинолон гетероциклы аналогично кверцетиндиоксигеназа, но считается, что они опосредуют радикальную реакцию молекулы дикислорода с карбанион на подложке (рисунок 5).[17] И HDO, и QDO принадлежат α / β гидролаза суперсемейство ферментов, хотя каталитические остатки в HDO и QDO, по-видимому, не выполняют ту же функцию, что и в остальных ферментах суперсемейства α / β гидролаз.[16]
Клиническое значение
Из-за разнообразия семейства диоксигеназ диоксигеназы имеют широкий спектр влияний в биологии:
- Триптофан-2,3-диоксигеназа (TDO) важен для регулирования уровней триптофан в организме и выражается в большом количестве опухолей человека.[18] Другая гемовая железозависимая диоксигеназа, IDO, также имеет отношение к здоровью человека, поскольку она участвует в воспалительных реакциях в контексте определенных заболеваний.[19] Поскольку он влияет на уровень как триптофана, так и кинуренин, IDO также участвует во влиянии на системы, связанные с депрессией у людей.[20]
- Алькаптонурия это генетическое заболевание, которое приводит к дефициту гомогентизат 1,2-диоксигеназа, который отвечает за катализирование образования 4-малейлацетоацетат из гомогентизат.[21] Накопление гомогентизиновой кислоты может привести к повреждению сердечного клапана, камням в почках и повреждению хрящей в организме.[22]
- Нейродегенерация, связанная с пантотенаткиназой (PKAN) - это аутосомно-рецессивный нарушение, которое может привести к развитию гранул железа и Тела Леви в нейроны. Исследование показало, что у пациентов с диагнозом PKAN было обнаружено увеличение цистеин уровни в бледный шар как следствие цистеиндиоксигеназа дефицит.[23] У пациентов с PKAN часто появляются симптомы слабоумие и часто умирают в раннем возрасте во взрослом возрасте.
- При репарации ДНК Fe (II) / 2-оксоглутарат-зависимая диоксигеназа AlkB, действует в окислительном удалении повреждений ДНК алкилирования. Неспособность удалить повреждение алкилирования ДНК может привести к цитотоксичности или мутагенезу во время репликации ДНК.
- Циклооксигеназы (COX), которые отвечают за формирование простаноиды в человеческом теле, являются целью многих НПВП обезболивающие.[10] Подавление ЦОГ приводит к уменьшению воспаления и имеет обезболивающий эффект из-за пониженного уровня синтеза простагландина и тромбоксана.
Рекомендации
- ^ а б Leitgeb S, Nidetzky B (декабрь 2008 г.). «Структурное и функциональное сравнение 2-His-1-карбоксилата и 3-His металлоцентров в негемных железо (II) -зависимых ферментах». Сделки Биохимического Общества. 36 (Pt 6): 1180–6. Дои:10.1042 / BST0361180. PMID 19021520.
- ^ а б c d е ж Абу-Омар М.М., Лоайза А., Хонцеас Н. (июнь 2005 г.). «Механизмы реакций мононуклеарных негемовых оксигеназ железа». Химические обзоры. 105 (6): 2227–52. Дои:10.1021 / cr040653o. PMID 15941213.
- ^ а б c d е ж Самуэль де Виссер; Девеш Кумар (2011). Железосодержащие ферменты универсальные катализаторы реакций гидроксилирования в природе. Королевское химическое общество. ISBN 978-1-84973-298-7.
- ^ а б c d е ж грамм час Fetzner S (апрель 2012 г.). «Кольцо-расщепляющие диоксигеназы с купиновой складкой». Прикладная и экологическая микробиология. 78 (8): 2505–14. Дои:10.1128 / AEM.07651-11. ЧВК 3318818. PMID 22287012.
- ^ Стипанук MH, Симмонс CR, Karplus PA, Dominy JE (июнь 2011 г.). «Тиолдиоксигеназы: уникальные семейства купиновых белков». Аминокислоты. 41 (1): 91–102. Дои:10.1007 / s00726-010-0518-2. ЧВК 3136866. PMID 20195658.
- ^ а б Соломон Э.И., Брунольд Т.С., Дэвис М.И., Кемсли Дж. Н., Ли С. К., Ленерт Н. и др. (Январь 2000 г.). «Геометрическая и электронная структура / взаимосвязь функций в негемовых ферментах железа». Химические обзоры. 100 (1): 235–350. Дои:10.1021 / cr9900275. PMID 11749238.
- ^ а б c Багг Т.Д., Рамасвами С. (апрель 2008 г.). «Негемовые железозависимые диоксигеназы: раскрытие каталитических механизмов сложных ферментативных окислений». Современное мнение в области химической биологии. 12 (2): 134–40. Дои:10.1016 / j.cbpa.2007.12.007. PMID 18249197.
- ^ а б Кребс С., Галонич Фухимори Д., Уолш К. Т., Боллинджер Дж. М. (июль 2007 г.). «Негемовые промежуточные соединения Fe (IV) -оксо». Отчеты о химических исследованиях. 40 (7): 484–92. Дои:10.1021 / ar700066p. ЧВК 3870002. PMID 17542550.
- ^ а б c Ефимов И., Басран Дж., Текрей С.Дж., Ханда С., Моват К.Г., Ворон Е.Л. (апрель 2011 г.). «Структура и механизм реакции в гем-диоксигеназах». Биохимия. 50 (14): 2717–24. Дои:10.1021 / bi101732n. ЧВК 3092302. PMID 21361337.
- ^ а б Соно М., Роуч МП, Колтер Э.Д., Доусон Дж. Х. (ноябрь 1996 г.). «Гемсодержащие оксигеназы». Химические обзоры. 96 (7): 2841–2888. Дои:10.1021 / cr9500500. PMID 11848843.
- ^ а б Thackray SJ, Mowat CG, Chapman SK (декабрь 2008 г.). «Изучение механизма действия триптофан-2,3-диоксигеназы». Сделки Биохимического Общества. 36 (Pt 6): 1120–3. Дои:10.1042 / BST0361120. ЧВК 2652831. PMID 19021508.
- ^ а б c d Шааб М.Р., Барни Б.М., Франциско В.А. (январь 2006 г.). «Кинетические и спектроскопические исследования кверцетин-2,3-диоксигеназы из Bacillus subtilis». Биохимия. 45 (3): 1009–16. Дои:10.1021 / bi051571c. PMID 16411777.
- ^ «Единственная супероксиддисмутаза Rhodobacter capsulatus представляет собой камбиалистический марганецсодержащий фермент». Jb.asm.org. Получено 2014-03-11.
- ^ а б Марони MJ, Ciurli S (апрель 2014 г.). "Никелевые ферменты без окисления". Химические обзоры. 114 (8): 4206–28. Дои:10.1021 / cr4004488. ЧВК 5675112. PMID 24369791.
- ^ а б Бур Дж. Л., Малруни С. Б., Хаусингер Р. П. (февраль 2014 г.). «Никельзависимые металлоферменты». Архивы биохимии и биофизики. 544: 142–52. Дои:10.1016 / j.abb.2013.09.002. ЧВК 3946514. PMID 24036122.
- ^ а б Fetzner S (ноябрь 2002 г.). «Оксигеназы без потребности в кофакторах или ионах металлов». Прикладная микробиология и биотехнология. 60 (3): 243–57. Дои:10.1007 / s00253-002-1123-4. PMID 12436305.
- ^ Bugg TD (сентябрь 2003 г.). «Ферменты диоксигеназы: каталитические механизмы и химические модели». Тетраэдр. 59 (36): 7075–7101. Дои:10.1016 / S0040-4020 (03) 00944-X.
- ^ Pilotte L, Larrieu P, Stroobant V, Colau D, Dolusic E, Frédérick R, De Plaen E, Uyttenhove C, Wouters J, Masereel B, Van den Eynde BJ (февраль 2012 г.). «Обращение к опухолевой иммунной резистентности путем ингибирования триптофан-2,3-диоксигеназы». Труды Национальной академии наук Соединенных Штатов Америки. 109 (7): 2497–502. Дои:10.1073 / pnas.1113873109. ЧВК 3289319. PMID 22308364.
- ^ Мураками Ю., Хоши М., Имамура Ю., Ариока Ю., Ямамото Ю., Сайто К. (2013). «Замечательная роль индоламин-2,3-диоксигеназы и метаболитов триптофана в инфекционных заболеваниях: потенциальная роль в воспалительных заболеваниях, опосредованных макрофагами». Медиаторы воспаления. 2013: 391984. Дои:10.1155/2013/391984. ЧВК 3588179. PMID 23476103.
- ^ Sublette ME, Postolache TT (сентябрь 2012 г.). «Нейровоспаление и депрессия: роль индоламин-2,3-диоксигеназы (IDO) как молекулярный путь». Психосоматическая медицина. 74 (7): 668–72. Дои:10.1097 / PSY.0b013e318268de9f. PMID 22923699.
- ^ Воет Д., Воет Дж. Г. (2011). Биохимия (4-е изд.). Хобокен, Нью-Джерси: Джон Уайли и сыновья. п. 1045. ISBN 0470917458.
- ^ Форнпхуткул С., Интрон В.Дж., Перри МБ, Бернардини И., Мерфи М.Д., Фицпатрик Д.Л., Андерсон П.Д., Хейзинг М., Аникстер Ю., Гербер Л.Х., Гал В.А. (декабрь 2002 г.). «Естественная история алкаптонурии». Медицинский журнал Новой Англии. 347 (26): 2111–21. Дои:10.1056 / NEJMoa021736. PMID 12501223.
- ^ Перри Т.Л., Норман М.Г., Йонг В.В., Уайтинг С., Крайтон Дж. Ю, Хансен С., Киш С. Дж. (Октябрь 1985 г.). «Болезнь Халлервордена-Шпатца: накопление цистеина и дефицит цистеиндиоксигеназы в бледном шаре». Анналы неврологии. 18 (4): 482–9. Дои:10.1002 / ana.410180411. PMID 4073841.