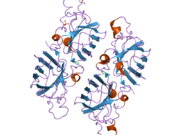SOD1 - SOD1

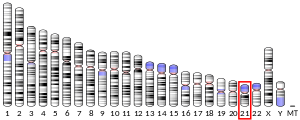




Супероксиддисмутаза [Cu-Zn] также известный как супероксиддисмутаза 1 или же SOD1 является фермент что у людей кодируется SOD1 ген, расположен на хромосома 21. SOD1 - один из трех человеческих супероксиддисмутазы.[5][6] Это замешано в апоптоз и семейный боковой амиотрофический склероз.[6]
Структура
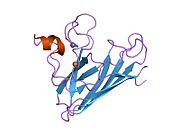
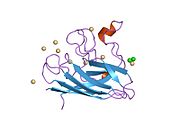
SOD1 - 32 кДа гомодимер который образует β-бочку и содержит внутримолекулярную дисульфидную связь и биядерный сайт Cu / Zn в каждой субъединице. Этот сайт Cu / Zn содержит ион меди и цинка и отвечает за катализ непропорциональность из супероксид к пероксид водорода и диоксид кислорода.[7][8] Процесс созревания этого белка сложен и до конца не изучен, включая избирательное связывание ионов меди и цинка, образование внутрисубъединицы. дисульфидная связь между Cys-57 и Cys-146 и димеризацией двух субъединиц. Медный шаперон для Sod1 (CCS) облегчает внедрение меди и окисление дисульфидов. Хотя SOD1 синтезируется в цитозоле и может там созревать, часть экспрессируемого и еще незрелого SOD1, нацеленного на митохондрии, должна быть вставлена в межмембранное пространство. Там он образует дисульфидную связь, но не металлизацию, необходимую для ее созревания.[8] Зрелый белок очень стабилен,[9] но нестабилен в формах, не содержащих металлов и восстановленных дисульфидом.[7][8][9] Это проявляется in vitro, так как потеря ионов металлов приводит к увеличению агрегации SOD1, а также в моделях болезней, где наблюдается низкая металлизация нерастворимого SOD1. Более того, восстановленные цистеины на поверхности могут участвовать в дисульфидных сшивание и, таким образом, агрегирование.[7]
Функция
SOD1 связывает ионы меди и цинка и является одной из трех супероксиддисмутаз, ответственных за разрушение свободных супероксид радикалы в организме. Закодированный изофермент растворимый цитоплазматический и митохондриальный белок межмембранного пространства, действующий как гомодимер для превращения встречающихся в природе, но вредных супероксидных радикалов в молекулярный кислород и пероксид водорода.[8][10] Затем перекись водорода может расщепляться другим ферментом, называемым каталазой.
Постулируется, что SOD1 локализовать к внешняя митохондриальная мембрана (OMM), где будут генерироваться супероксидные анионы, или межмембранное пространство. Точные механизмы его локализации остаются неизвестными, но его агрегация с OMM приписывается его ассоциации с BCL-2. SOD1 дикого типа продемонстрировал антиапоптотические свойства в невральных культурах, в то время как мутантный SOD1, как было замечено, способствует апоптозу в митохондриях спинного мозга, но не в печень митохондрии, хотя он одинаково выражен в обоих. Две модели предполагают, что SOD1 ингибирует апоптоз, взаимодействуя с BCL-2 белки или сами митохондрии.[6]
Клиническое значение
Роль в окислительном стрессе
В частности, SOD1 играет ключевую роль в активные формы кислорода (ROS) высвобождение во время окислительного стресса в результате ишемии-реперфузионного повреждения, особенно в миокарде как часть острое сердечно-сосудистое заболевание (также известный как ишемическая болезнь сердца ). Ишемическая болезнь сердца, возникающая в результате окклюзия одного из главных коронарные артерии, в настоящее время по-прежнему является основной причиной болезненность и смертность в западном обществе.[11][12] Во время ишемической реперфузии высвобождение АФК вносит существенный вклад в повреждение и гибель клеток посредством прямого воздействия на клетки, а также посредством сигналов апоптоза. Известно, что SOD1 обладает способностью ограничивать пагубные эффекты ROS. Таким образом, SOD1 важен благодаря своим кардиозащитным эффектам.[13] Кроме того, SOD1 участвует в кардиопротекции против ишемического реперфузионного повреждения, например, во время ишемическое прекондиционирование сердца.[14] Хотя известно, что большой всплеск АФК приводит к повреждению клеток, умеренное высвобождение АФК из митохондрий, которое происходит во время несмертельных коротких эпизодов ишемии, может играть значительную пусковую роль в путях передачи сигнала ишемического прекондиционирования, приводящего к снижению повреждение клеток. Было даже замечено, что во время этого высвобождения ROS, SOD1 играет важную роль, регулируя передачу сигналов апоптоза и гибель клеток.
В одном исследовании делеции гена были зарегистрированы в двух семейных случаях: кератоконус.[15] У мышей, лишенных SOD1, наблюдается повышенная возрастная потеря мышечной массы (саркопения ), раннее развитие катаракта, дегенерация желтого пятна, инволюция тимуса, гепатоцеллюлярная карцинома, и сокращенная продолжительность жизни.[16] Исследования показывают, что повышенный уровень SOD1 может быть биомаркером хронического токсичность тяжелых металлов у женщин с длительным стоматологическая амальгама начинки.[17]
Боковой амиотрофический склероз (болезнь Лу Герига)
Мутации (на сегодняшний день идентифицировано более 150) в этом гене связаны с семейными боковой амиотрофический склероз.[18][19][20] Однако несколько свидетельств также показывают, что SOD1 дикого типа в условиях клеточного стресса участвует в значительной части спорадических случаев БАС, которые составляют 90% пациентов с БАС.[21]Наиболее частые мутации: A4V (в США) и H46R (Япония). Только в Исландии SOD1-G93S был найден. Наиболее изученная модель мыши с БАС - это G93A. Сообщалось о редких вариантах транскрипта для этого гена.[10]
Практически все известные мутации SOD1, вызывающие БАС, действуют в доминирующий мода; одной мутантной копии гена SOD1 достаточно, чтобы вызвать заболевание. Точный молекулярный механизм (или механизмы), с помощью которого мутации SOD1 вызывают заболевание, неизвестен. Похоже, это какое-то токсическое усиление функции,[20] поскольку многие связанные с заболеванием мутанты SOD1 (включая G93A и A4V) сохраняют ферментативную активность, а у мышей с нокаутом Sod1 не развивается БАС (хотя они действительно проявляют сильную возрастную дистальную моторную невропатию).
ALS это нейродегенеративное заболевание характеризуется избирательной потерей двигательные нейроны вызывая мышечная атрофия. В Окисление ДНК товар 8-OHdG является признанным маркером окислительное повреждение ДНК. 8-OHdG накапливается в митохондрии позвоночника двигательные нейроны людей с БАС.[22] В трансгенный У мышей с БАС, несущих мутантный ген SOD1, 8-OHdG также накапливается в митохондриальная ДНК мотонейронов спинного мозга.[23] Эти данные позволяют предположить, что окислительное повреждение митохондриальной ДНК двигательных нейронов из-за изменения SOD1 может быть важным фактором в этиологии БАС.
Мутация A4V
A4V (аланин в кодоне 4 изменен на валин ) является наиболее распространенной мутацией, вызывающей БАС, в популяции США: примерно 50% пациентов с СОД1-БАС несут мутацию A4V.[24][25][26] Примерно 10 процентов всех семейных случаев БАС в США вызваны гетерозиготными мутациями A4V в SOD1. Мутация редко встречается за пределами Америки.
Недавно было подсчитано, что мутация A4V произошла 540 поколений (~ 12 000 лет) назад. Гаплотип, окружающий мутацию, предполагает, что мутация A4V возникла у азиатских предков коренных американцев, которые достигли Америки через Берингов пролив.[27]
Мутант A4V относится к WT-подобным мутантам. Пациенты с мутациями A4V демонстрируют различный возраст начала, но всегда очень быстрое течение болезни, средняя выживаемость после начала составляет 1,4 года (по сравнению с 3-5 годами с другими доминантными мутациями SOD1, а в некоторых случаях, таких как H46R, значительно дольше). Эта выживаемость значительно короче, чем у немутантного SOD1-связанного БАС.
Мутация H46R
H46R (гистидин в кодоне 47 изменен на аргинин ) является наиболее распространенной мутацией, вызывающей БАС, в популяции Японии, причем около 40% японских пациентов с СОД1-БАС несут эту мутацию. H46R вызывает глубокую потерю связывания меди в активном центре SOD1, и, как таковой, H46R ферментативно неактивен. Болезнь, вызванная этой мутацией, длится очень долго, обычно от начала заболевания до смерти более 15 лет.[28] Мышиные модели с этой мутацией не демонстрируют классическую патологию митохондриальной вакуолизации, наблюдаемую у мышей с БАС G93A и G37R, и, в отличие от мышей G93A, дефицит основного митохондриального антиоксидантного фермента, SOD2, не влияет на течение болезни.[28]
Мутация G93A
G93A (глицин 93 заменен на аланин) - сравнительно редкая мутация, но она изучается очень интенсивно, поскольку это была первая мутация, смоделированная на мышах. G93A - это мутация псевдо-WT, которая не влияет на активность фермента.[26] Из-за доступности мыши G93A от Лаборатория Джексона, на этой модели было проведено множество исследований потенциальных мишеней лекарств и механизмов токсичности. Как минимум один частный исследовательский институт (Институт развития терапии БАС ) проводит крупномасштабные анализы наркотиков исключительно на этой модели мыши. Являются ли результаты специфическими для G93A или применимыми ко всем БАС, вызывающим мутации SOD1, в настоящее время неизвестно. Утверждалось, что определенные патологические особенности мыши G93A обусловлены артефактами сверхэкспрессии, в частности, артефактами, связанными с митохондриальной вакуолизацией (мышь G93A, обычно используемая из лаборатории Джексона, имеет более 20 копий гена SOD1 человека).[29] По крайней мере, одно исследование показало, что некоторые особенности патологии являются идиосинкразическими для G93A и не могут быть экстраполированы на все мутации, вызывающие БАС.[28] Дальнейшие исследования показали, что патогенез моделей G93A и H46R четко различается; некоторые лекарства и генетические вмешательства, которые очень полезны / вредны в одной модели, имеют либо противоположный эффект, либо не действуют в другой.[30][31][32]
Синдром Дауна
Синдром Дауна (DS) вызвано утроение хромосомы 21. Окислительный стресс считается важным фактором, лежащим в основе патологий, связанных с СД. Окислительный стресс, по-видимому, связан с троектированием и повышенной экспрессией гена SOD1, расположенного в хромосоме 21. Повышенная экспрессия SOD1, вероятно, вызывает повышенную продукцию пероксид водорода что приводит к увеличению клеточного повреждения.
Уровни 8-OHdG в ДНК лиц с DS, измеряемых в слюна, оказались значительно выше, чем в контрольных группах.[33] Уровни 8-OHdG также были увеличены в лейкоциты людей с DS по сравнению с контролем.[34] Эти данные свидетельствуют о том, что окислительное повреждение ДНК может приводить к некоторым клиническим проявлениям СД.
Взаимодействия
Было показано, что SOD1 взаимодействовать с CCS[35] и Bcl-2.[36][37][38][39]
Рекомендации
- ^ а б c ГРЧ38: Ансамбль выпуск 89: ENSG00000142168 - Ансамбль, Май 2017
- ^ а б c GRCm38: выпуск Ensembl 89: ENSMUSG00000022982 - Ансамбль, Май 2017
- ^ "Справочник человека по PubMed:". Национальный центр биотехнологической информации, Национальная медицинская библиотека США.
- ^ "Ссылка на Mouse PubMed:". Национальный центр биотехнологической информации, Национальная медицинская библиотека США.
- ^ Милани П., Гальярди С., Кова Е., Середа С. (2011). «Регуляция транскрипции и посттранскрипции SOD1 и ее потенциальные последствия при БАС». Neurology Research International. 2011: 1–9. Дои:10.1155/2011/458427. ЧВК 3096450. PMID 21603028.
- ^ а б c Розен Д.Р., Сиддик Т., Паттерсон Д., Фиглевич Д.А., Сапп П., Хентати А., Дональдсон Д., Гото Дж., О'Реган Д.П., Дэн Х.Х. (март 1993 г.). «Мутации в гене супероксиддисмутазы Cu / Zn связаны с семейным боковым амиотрофическим склерозом». Природа. 362 (6415): 59–62. Bibcode:1993Натура 362 ... 59р. Дои:10.1038 / 362059a0. PMID 8446170. S2CID 265436.
- ^ а б c Estácio SG, Leal SS, Cristóvão JS, Faísca PF, Gomes CM (февраль 2015 г.). «Связывание кальция с привратными остатками, фланкирующими склонные к агрегации сегменты, лежит в основе нефибриллярных амилоидных черт в супероксиддисмутазе 1 (SOD1)». Biochimica et Biophysica Acta (BBA) - Белки и протеомика. 1854 (2): 118–26. Дои:10.1016 / j.bbapap.2014.11.005. PMID 25463043.
- ^ а б c d Sea K, Sohn SH, Durazo A, Sheng Y, Shaw BF, Cao X, Taylor AB, Whitson LJ, Holloway SP, Hart PJ, Cabelli DE, Gralla EB, Valentine JS (январь 2015 г.). «Понимание роли необычной дисульфидной связи в супероксиддисмутазе медь-цинк». Журнал биологической химии. 290 (4): 2405–18. Дои:10.1074 / jbc.M114.588798. ЧВК 4303690. PMID 25433341.
- ^ а б Харе С.Д., Каплоу М., Дохолян Н.В. (октябрь 2004 г.). «Константы скорости и равновесия для многоступенчатой последовательности реакций агрегации супероксиддисмутазы при боковом амиотрофическом склерозе». Труды Национальной академии наук Соединенных Штатов Америки. 101 (42): 15094–9. Bibcode:2004ПНАС..10115094К. Дои:10.1073 / pnas.0406650101. ЧВК 524068. PMID 15475574.
- ^ а б «Ген Entrez: супероксиддисмутаза 1 SOD1, растворимая (боковой амиотрофический склероз 1 (взрослый))».
- ^ Мюррей CJ, Лопес AD (май 1997 г.). «Альтернативные прогнозы смертности и инвалидности по причинам 1990-2020: Исследование глобального бремени болезней». Ланцет. 349 (9064): 1498–504. Дои:10.1016 / S0140-6736 (96) 07492-2. PMID 9167458. S2CID 10556268.
- ^ Браунвальд Э., Клонер Р.А. (ноябрь 1985 г.). «Реперфузия миокарда: палка о двух концах?». Журнал клинических исследований. 76 (5): 1713–9. Дои:10.1172 / JCI112160. ЧВК 424191. PMID 4056048.
- ^ Маслов Л.Н., Нарыжная Н.В., Подоксенов Ю.К., Прокудина Е.С., Горбунов А.С., Чжан И., Пе Ж.М. (январь 2015). «[Активные формы кислорода являются триггерами и медиаторами повышения толерантности сердца к воздействию ишемии-реперфузии]». Российский Физиологический Журнал Имени И.М. Сеченова / Российская Академия Наук.. 101 (1): 3–24. PMID 25868322.
- ^ Liem DA, Honda HM, Zhang J, Woo D, Ping P (декабрь 2007 г.). «Прошлый и настоящий курс кардиопротекции против ишемического реперфузионного повреждения». Журнал прикладной физиологии. 103 (6): 2129–36. Дои:10.1152 / japplphysiol.00383.2007. PMID 17673563.
- ^ Udar N, Atilano SR, Brown DJ, Holguin B, Small K, Nesburn AB, Kenney MC (август 2006 г.). «SOD1: ген-кандидат кератоконуса». Исследовательская офтальмология и визуализация. 47 (8): 3345–51. Дои:10.1167 / iovs.05-1500. PMID 16877401.
- ^ Muller FL, Lustgarten MS, Jang Y, Richardson A, Van Remmen H (август 2007 г.). «Тенденции в теориях окислительного старения». Свободная радикальная биология и медицина. 43 (4): 477–503. Дои:10.1016 / j.freeradbiomed.2007.03.034. PMID 17640558.
- ^ Cabaña-Muñoz ME, Parmigiani-Izquierdo JM, Bravo-González LA, Kyung HM, Merino JJ (июнь 2015 г.). «Повышенный уровень цинка / глутатиона и более высокая активность супероксиддисмутазы-1 как биомаркеры окислительного стресса у женщин с длительными пломбами из зубной амальгамы: корреляция между уровнями ртути / алюминия (в волосах) и антиоксидантными системами в плазме». PLOS ONE. 10 (6): e0126339. Bibcode:2015PLoSO..1026339C. Дои:10.1371 / journal.pone.0126339. ЧВК 4468144. PMID 26076368.
- ^ Conwit RA (декабрь 2006 г.). «Предотвращение семейного БАС: клиническое испытание возможно, но оправдано ли испытание эффективности?». Журнал неврологических наук. 251 (1–2): 1–2. Дои:10.1016 / j.jns.2006.07.009. PMID 17070848. S2CID 33105812.
- ^ Аль-Чалаби А., Ли П. Н. (август 2000 г.). «Последние достижения в области бокового амиотрофического склероза». Текущее мнение в неврологии. 13 (4): 397–405. Дои:10.1097/00019052-200008000-00006. PMID 10970056. S2CID 21577500.
- ^ а б Редлер Р.Л., Дохолян Н.В. (01.01.2012). «Комплексная молекулярная биология бокового амиотрофического склероза (БАС)». Молекулярная биология нейродегенеративных заболеваний. Прогресс в молекулярной биологии и трансляционной науке. 107. С. 215–62. Дои:10.1016 / B978-0-12-385883-2.00002-3. ISBN 9780123858832. ЧВК 3605887. PMID 22482452.
- ^ Гальярди С., Кова Е., Давин А., Гуарески С., Абель К., Алвиси Е., Лафоренца Ю., Гидони Р., Кашман Дж. Р., Серони М., Середа С. (август 2010 г.). «Экспрессия мРНК SOD1 при спорадическом боковом амиотрофическом склерозе». Нейробиология болезней. 39 (2): 198–203. Дои:10.1016 / j.nbd.2010.04.008. PMID 20399857. S2CID 207065284.
- ^ Кикучи Х., Фурута А., Нисиока К., Судзуки С.О., Накабеппу Ю., Иваки Т. (апрель 2002 г.). «Нарушение митохондриальных ферментов репарации ДНК против накопления 8-оксогуанина в моторных нейронах спинного мозга при боковом амиотрофическом склерозе». Acta Neuropathol. 103 (4): 408–14. Дои:10.1007 / s00401-001-0480-х. PMID 11904761. S2CID 2102463.
- ^ Варита Х, Хаяси Т., Мураками Т., Манабе Й., Абэ К. (апрель 2001 г.). «Окислительное повреждение митохондриальной ДНК в спинномозговых мотонейронах трансгенных мышей с БАС». Brain Res. Мол. Brain Res. 89 (1–2): 147–52. Дои:10.1016 / S0169-328X (01) 00029-8. PMID 11311985.
- ^ Розен Д.Р., Боулинг А.С., Паттерсон Д., Усдин Т.Б., Сапп П., Мези Э., Маккенна-Ясек Д., О'Реган Дж., Рахмани З., Ферранте Р.Дж. (июнь 1994 г.). «Частая мутация супероксиддисмутазы-1 от ala 4 до val связана с быстро прогрессирующим семейным боковым амиотрофическим склерозом». Молекулярная генетика человека. 3 (6): 981–7. Дои:10,1093 / hmg / 3,6,981. PMID 7951249.
- ^ Cudkowicz ME, McKenna-Yasek D, Sapp PE, Chin W, Geller B, Hayden DL, Schoenfeld DA, Hosler BA, Horvitz HR, Brown RH (февраль 1997 г.). «Эпидемиология мутаций супероксиддисмутазы при боковом амиотрофическом склерозе». Анналы неврологии. 41 (2): 210–21. Дои:10.1002 / ana.410410212. PMID 9029070. S2CID 25595595.
- ^ а б Валентин Дж. С., Харт П. Дж. (Апрель 2003 г.). «Неправильно свернутый CuZnSOD и боковой амиотрофический склероз». Труды Национальной академии наук Соединенных Штатов Америки. 100 (7): 3617–22. Bibcode:2003ПНАС..100.3617В. Дои:10.1073 / pnas.0730423100. ЧВК 152971. PMID 12655070.
- ^ Брум В.Дж., Джонсон Д.В., Аувартер К.Э., Яфрат А.Дж., Расс С., Аль-Чалаби А., Сапп П.С., Маккенна-Ясек Д., Андерсен П.М., Браун Р.Х. (январь 2008 г.). «SOD1A4V-опосредованный БАС: отсутствие тесно связанного гена-модификатора и происхождение в Азии». Письма о неврологии. 430 (3): 241–5. Дои:10.1016 / j.neulet.2007.11.004. PMID 18055113. S2CID 46282375.
- ^ а б c Мюллер Флорида, Лю Ю., Джерниган А., Борчелт Д., Ричардсон А., Ван Реммен Х. (сентябрь 2008 г.). «Дефицит MnSOD по-разному влияет на прогрессирование заболевания в двух разных моделях мутантных мышей с БАС». Мышцы и нервы. 38 (3): 1173–83. Дои:10.1002 / mus.21049. PMID 18720509. S2CID 23971601.
- ^ Бергемальм Д., Йонссон П.А., Граффмо К.С., Андерсен П.М., Бреннстрем Т., Реммарк А., Марклунд С.Л. (апрель 2006 г.). «Перегрузка стабильных и исключение нестабильных вариантов супероксиддисмутазы-1 человека в митохондриях моделей бокового амиотрофического склероза мышей». Журнал неврологии. 26 (16): 4147–54. Дои:10.1523 / JNEUROSCI.5461-05.2006. ЧВК 6673995. PMID 16624935.
- ^ Пан Л., Йошии Й, Отомо А., Огава Х, Ивасаки Й, Шан Х. Ф., Хадано С. (2012). «Различные мутанты супероксиддисмутазы меди и цинка человека, SOD1G93A и SOD1H46R, оказывают различное вредное воздействие на общий фенотип у мышей». PLOS ONE. 7 (3): e33409. Bibcode:2012PLoSO ... 733409P. Дои:10.1371 / journal.pone.0033409. ЧВК 3306410. PMID 22438926.
- ^ Бхаттачарья А., Боков А., Мюллер Флорида, Джерниган А. Л., Маслин К., Диаз В., Ричардсон А., Ван Реммен Х (август 2012 г.). «Ограничение диеты, но не рапамицин, увеличивает время начала заболевания и выживаемость мышиной модели БАС H46R / H48Q». Нейробиология старения. 33 (8): 1829–32. Дои:10.1016 / j.neurobiolaging.2011.06.002. PMID 21763036. S2CID 11227242.
- ^ Варгас М.Р., Джонсон Д.А., Джонсон Дж.А. (сентябрь 2011 г.). «Снижение глутатиона ускоряет неврологический дефицит и митохондриальную патологию на модели мышей с семейным БАС-связанным hSOD1 (G93A)». Нейробиология болезней. 43 (3): 543–51. Дои:10.1016 / j.nbd.2011.04.025. ЧВК 3139005. PMID 21600285.
- ^ Komatsu T, Duckyoung Y, Ito A, Kurosawa K, Maehata Y, Kubodera T, Ikeda M, Lee MC (сентябрь 2013 г.). «Повышенные биомаркеры оксидативного стресса в слюне пациентов с синдромом Дауна». Arch. Оральный Биол. 58 (9): 1246–50. Дои:10.1016 / j.archoralbio.2013.03.017. PMID 23714170.
- ^ Паллардо Ф.В., Деган П., д'Искья М., Келли Ф. Дж., Заттерале А., Кальцоне Р., Кастелло Дж., Фернандес-Дельгадо Р., Данстер С., Льорет А., Манини П., Пизанти М. А., Вуттариелло Е., Пагано Г. (август 2006 г.). «Множественные доказательства прооксидантного состояния в раннем возрасте у пациентов с синдромом Дауна». Биогеронтология. 7 (4): 211–20. Дои:10.1007 / s10522-006-9002-5. PMID 16612664. S2CID 13657691.
- ^ Касарено Р.Л., Вагонер Д., Гитлин Д.Д. (сентябрь 1998 г.). «Шаперон меди CCS напрямую взаимодействует с супероксиддисмутазой меди / цинка». Журнал биологической химии. 273 (37): 23625–8. Дои:10.1074 / jbc.273.37.23625. PMID 9726962.
- ^ Пасинелли П., Белфорд М.Э., Леннон Н., Бакскаи Б.Дж., Хайман Б.Т., Тротти Д., Браун Р.Х. (июль 2004 г.). «Связанные с амиотрофическим боковым склерозом мутантные белки SOD1 связываются и агрегируются с Bcl-2 в митохондриях спинного мозга». Нейрон. 43 (1): 19–30. Дои:10.1016 / j.neuron.2004.06.021. PMID 15233914. S2CID 18141051.
- ^ Кова Е., Гирольди А., Гуарески С., Мадзини Дж., Гальярди С., Давин А., Бьянки М., Черони М., Середа С. (октябрь 2010 г.). «G93A SOD1 изменяет клеточный цикл в клеточной модели бокового амиотрофического склероза». Сотовая связь. 22 (10): 1477–84. Дои:10.1016 / j.cellsig.2010.05.016. PMID 20561900.
- ^ Cereda C, Cova E, Di Poto C, Galli A, Mazzini G, Corato M, Ceroni M (ноябрь 2006 г.). «Влияние оксида азота на лимфоциты пациентов со спорадическим боковым амиотрофическим склерозом: токсическая или защитная роль?». Неврологические науки. 27 (5): 312–6. Дои:10.1007 / s10072-006-0702-z. PMID 17122939. S2CID 25059353.
- ^ Cova E, Cereda C, Galli A, Curti D, Finotti C, Di Poto C, Corato M, Mazzini G, Ceroni M (май 2006 г.). «Модифицированная экспрессия белков Bcl-2 и SOD1 в лимфоцитах от пациентов со спорадическим БАС». Письма о неврологии. 399 (3): 186–90. Дои:10.1016 / j.neulet.2006.01.057. PMID 16495003. S2CID 26076370.
дальнейшее чтение
- де Беллерош Дж., Оррелл Р., Кинг А. (ноябрь 1995 г.). «Семейный боковой амиотрофический склероз / заболевание двигательных нейронов (FALS): обзор текущих событий». Журнал медицинской генетики. 32 (11): 841–7. Дои:10.1136 / jmg.32.11.841. ЧВК 1051731. PMID 8592323.
- Серони М., Курти Д., Алимонти Д. (2002). «Боковой амиотрофический склероз и ген SOD1: обзор». Функциональная неврология. 16 (4 Suppl): 171–80. PMID 11996514.
- Зелко И.Н., Мариани Т.Дж., Фольц Р.Дж. (август 2002 г.). «Мультигенное семейство супероксиддисмутазы: сравнение структур, эволюции и экспрессии генов CuZn-SOD (SOD1), Mn-SOD (SOD2) и EC-SOD (SOD3)». Свободная радикальная биология и медицина. 33 (3): 337–49. Дои:10.1016 / S0891-5849 (02) 00905-X. PMID 12126755.
- Hadano S (июнь 2002 г.). «[Гены, вызывающие семейный боковой амиотрофический склероз]». Сэйкагаку. Журнал Японского биохимического общества. 74 (6): 483–9. PMID 12138710.
- Нур Р., Миттал С., Икбал Дж. (Сентябрь 2002 г.). «Супероксиддисмутаза - применение и актуальность при заболеваниях человека». Монитор медицинских наук. 8 (9): RA210–5. PMID 12218958.
- Поттер С.З., Валентайн Д.С. (апрель 2003 г.). «Загадочная роль супероксиддисмутазы цинка и меди в боковом амиотрофическом склерозе (болезнь Лу Герига)». Журнал биологической неорганической химии. 8 (4): 373–80. Дои:10.1007 / s00775-003-0447-6. PMID 12644909. S2CID 22820101.
- Ротилио Г., Аквилано К., Сириоло М.Р. (2004). «Взаимодействие Cu, Zn супероксиддисмутазы и синтазы оксида азота в нейродегенеративных процессах». IUBMB Life. 55 (10–11): 629–34. Дои:10.1080/15216540310001628717. PMID 14711010. S2CID 19518719.
- Джафари-Шлуп Х.Ф., Хорис Дж., Майё-Портас В., Хэнд С., Руло Г., Каму В. (январь 2004 г.). «[Аномалии гена супероксиддисмутазы 1 при семейном боковом амиотрофическом склерозе: корреляции фенотип / генотип. Французский опыт и обзор литературы]». Revue Neurologique. 160 (1): 44–50. Дои:10.1016 / S0035-3787 (04) 70846-2. PMID 14978393.
- Фарачи FM, Дидион SP (август 2004 г.). «Сосудистая защита: изоформы супероксиддисмутазы в стенке сосуда». Артериосклероз, тромбоз и биология сосудов. 24 (8): 1367–73. Дои:10.1161 / 01.ATV.0000133604.20182.cf. PMID 15166009.
- Gagliardi S, Ogliari P, Davin A, Corato M, Cova E, Abel K, Cashman JR, Ceroni M, Cereda C (август 2011 г.). «Уровни мРНК флавинсодержащей монооксигеназы повышаются в областях мозга также у мышей с мутантным SOD1». Исследования нейротоксичности. 20 (2): 150–8. Дои:10.1007 / s12640-010-9230-у. PMID 21082301. S2CID 21856030.
- Баттистини С., Риччи С., Лотти Е.М., Бениньи М., Гальярди С., Зукко Р., Бондавалли М., Марчелло Н., Черони М., Середа С. (июнь 2010 г.). «Тяжелый семейный БАС с новой мутацией экзона 4 (L106F) в гене SOD1». Журнал неврологических наук. 293 (1–2): 112–5. Дои:10.1016 / j.jns.2010.03.009. PMID 20385392. S2CID 24895265.
Галерея PDB | |
|---|---|
|