Врожденная мышечная дистрофия - Congenital muscular dystrophy
| Врожденная мышечная дистрофия | |
|---|---|
 | |
| Аутосомно-рецессивный тип обычно наследуется по типу CMD. | |
| Специальность | Неврология |
| Симптомы | Мышечная слабость[1] |
| Типы | 17 видов CMD[1] |
| Диагностический метод | NRI, EMG[2] |
| Уход | В настоящее время лекарства нет; следует контролировать сердечную функцию и функцию дыхания[3] |
Врожденные мышечные дистрофии находятся аутосомный рецессивно-унаследованный мышечные заболевания. Они группа гетерогенные расстройства характеризуется мышечной слабостью, которая присутствует при рождении, и различными изменениями биопсия мышц это колеблется от миопатический открыто дистрофический из-за возраста, в котором проводится биопсия.[1][4]
Признаки и симптомы
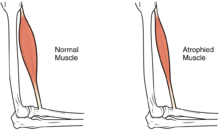
У большинства младенцев с ВМД будет наблюдаться прогрессирующая мышечная слабость или мышечное истощение (атрофия), хотя могут быть разные степени и симптомы серьезности прогрессирования. Слабость обозначена какгипотония или отсутствие мышечного тонуса, из-за чего ребенок может казаться нестабильным.[1][5]
Дети могут медлить со своими двигательные навыки; например, переворачиваться, сидеть или ходить, или даже не достичь этих жизненных вех. Некоторые из более редких форм CMD могут привести к значительным нарушениям обучаемости.[требуется медицинская цитата ]
Генетика
Генетика врожденной мышечной дистрофии является аутосомно-рецессивной, что означает, что для возникновения заболевания или признака должны присутствовать две копии аномального гена. В случае дефицита коллагена VI он является аутосомно-доминантным, что означает, что ребенок может унаследовать заболевание только от одной копии гена, присутствующей только у одного родителя.[1]
По данным Reed, 2009, распространенность врожденной мышечной дистрофии составляет 2,6-4,5 на 10 000 человек.[6] MDCIA, например, возникает из-за мутации в LAMA-2 ген и участвует в 6q2 хромосома.[7]
Механизм
Что касается механизма врожденной мышечной дистрофии, можно обнаружить, что, хотя существует много типов ВМД, гликозилирование из α-дистрогликан и изменения в этих генах, которые вовлечены, являются важной частью патофизиологии этого состояния.[8]
Диагностика

Для диагностики врожденной мышечной дистрофии проводятся следующие анализы / обследования:[2]
- Лабораторное исследование (СК уровни)
- Мышцы МРТ и особенно МРТ мышц всего тела недавно использовалась для описания мышечных аномалий у пациентов с первичным подтипом дефицита ламинина-α2 (мерозина) CMD.
- ЭМГ
- Генетическое тестирование
Классификация (разные виды врожденных мышечных дистрофий)
Подтипы врожденной мышечной дистрофии были установлены благодаря вариациям множества генов. Фенотип, а также, генотип классификации используются для установления подтипов в некоторой литературе.[1]
Обнаруживается, что врожденные мышечные дистрофии могут быть либо аутосомно-доминантный или же аутосомно-рецессивный с точки зрения наследования, хотя последнее встречается гораздо чаще[1]
Лица, страдающие врожденной мышечной дистрофией, относятся к одному из следующих типы:
- CMD с мозгом, также называемый заболевание мышц, глаз и головного мозга,[9] это редкая форма врожденной мышечной дистрофии (аутосомно-рецессивное заболевание), вызывающая отсутствие нормального мышечного тонуса, что может задерживать ходьбу из-за слабости, а также паралич глазных мышц и умственной отсталости, которая влияет на способ обработки информации людьми[9] Это вызвано мутацией в POMGNT1 ген.[9]
- CMD с сведенными (обращенными внутрь) большими пальцами. редкая форма CMD, вызывающая необратимое укорочение суставов пальцев ног и отсутствие мышечного тонуса, что может задерживать ходьбу из-за слабости человека. У человека с этой формой врожденной мышечной дистрофии может быть легкая мозжечок в некоторых случаях гипоплазия.[1]
- CMD / LGMD без MR-Первые годы жизни новорожденного начинаются со слабости, которая сказывается на двигательных способностях, ходьбе может быть совершена в подростковом возрасте, деформации и ригидности суставов. Суставы, шея и позвоночник; прогрессирующая кардиомиопатия в раннем возрасте; У человека могут быть нарушения сердечного ритма.[1]
- Большой связанный CMD в начале периода новорожденности младенец получает следующие проблемы; плохой мышечный тонус и слабая двигательная функция; человек представит с Интеллектуальная недееспособность и структура мозга, вероятно, будет ненормальной.[10]
- ВМД с атрофией мозжечка тяжелая гипоплазия мозжечка, плохой мышечный тонус, задержка двигательных этапов, отсутствие координации двигательных навыков, трудности с речью, непроизвольные движения и некоторые умственные нарушения. Кроме того, мышца биопсия не обнаруживает недостатков.[1]
- Синдром Уокера – Варбурга вначале прогрессирующая слабость и низкий мышечный тонус при рождении или в раннем младенчестве; мелкие мышцы; большинство больных детей не доживают до 3-х лет. Присутствуют проблемы со строением глаза, сопровождающиеся нарушением зрения.[11]
- CMD с первичной недостаточностью ламинина-α2 (мерозина) (MDC1A) интеллект у таких людей не затронут, проксимальные мышцы ослаблены и позвоночник ригиден, а также поражены дыхательные пути (с прогрессированием заболевания).[12]
- CMD / LGMD с MR слабость, деформация и ригидность суставов, присутствующие при рождении, плохой мышечный тонус, медленно прогрессирующий; люди могут иметь кисты мозжечка (или корковые проблемы), микроцефалия также может присутствовать. Может возникнуть аномальная гибкость, возможно искривление позвоночника.[1]
- CDG I (DPM3) некоторые из симптомов при рождении и на протяжении всей жизни младенца - слабость или плохой мышечный тонус. У пациента может быть кардиомиопатия (отсутствие обструкции оттока), повышение уровня сыворотки крови. креатинкиназа тоже может присутствовать. Могут присутствовать некоторые проблемы с IQ, а также слабость проксимальных мышц. Также следует отметить сокращение долихолфосфат манноза .[13]
- CDG I (DPM2) слабый мышечный тонус, начиная с первых недель жизни ребенка, у человека могут проявляться тяжелые неврологические физические характеристики, которые приводят к летальному исходу в раннем возрасте. Гипотония и миопатические лица могут присутствовать у таких людей, в то время как контрактуры суставов также могут присутствовать. Ну наконец то, миоклонические припадки может возникнуть в очень раннем возрасте (3 месяца).[14]
- CDG Ie (DPM1) при рождении у младенца будет слабость с поражением дыхательной системы, а также серьезные психические и психомоторные проблемы. К 3 годам ребенок может быть слепым из-за проблем с речью. Микроцефалия может возникнуть в раннем детстве, а также могут возникнуть судороги.[15]
- CMD с ригидностью позвоночника присутствующие при рождении могут иметь плохой мышечный тонус и слабость, сниженную дыхательную способность, мышцы могут быть деформированы, стабилизация в раннем возрасте или медленное снижение ригидности позвоночника, ограниченная подвижность для сгибания шеи и позвоночника, искривление позвоночника и прогрессирующая деформация и ригидность суставов, незначительные сердечный аномалии, нормальный интеллект.[16]

- CMD с неисправностью ламината A / C с первого года жизни ребенок слаб, у человека могут возникнуть проблемы с поднятием рук и головы. Может потребоваться назогастральный зонд, слабость конечностей и повышенный уровень креатинкиназы сыворотки. Человек может показать диафрагмальный манера дыхания.[17]
- Интегрин α7 слабость, которая присутствует при рождении, плохой мышечный тонус из-за поздней ходьбы, потеря мышечной ткани, умственная отсталость. Кроме того, был повышен уровень креатинкиназы.[18]
- Фукуяма CMD-в западных странах этот тип ВМД встречается редко, но в Японии он распространен. Воздействие этого заболевания на младенцев различается по степени тяжести. К ним относятся слабость мышечного тонуса в течение первого года, деформированные и жесткие суставы, искривления позвоночника, судороги, поражение глаз и умственная отсталость. У некоторых пациентов может быть ограничена подвижность при ходьбе.[19]
- CMD с дефицитом мерозина- слабость мышечного тонуса, присутствующая при рождении, спектра степени тяжести; может проявляться гипотония и плохое двигательное развитие. У большинства людей есть перивентрикулярный проблемы с белым веществом. Тем не мение, Интеллектуальная недееспособность в большинстве случаев встречается редко.[20]
- Мерозин-положительный CMD некоторые формы мерозин-положительного CMD: ранняя ригидность позвоночника, CMD с мышечной гипертрофией, CMD с мышечной гипертрофией и дыхательной недостаточностью.[21]

- Врожденная мышечная дистрофия Ульриха при рождении присутствует слабость и плохой мышечный тонус.
Дифференциальная диагностика
DDx врожденной мышечной дистрофии у пораженного человека выглядит следующим образом (также существуют ненервно-мышечные генетические состояния.[23]):[2]
- Метаболические миопатии
- Дистрофинопатии
- Мышечная дистрофия Эмери-Дрейфуса
Управление

Что касается лечения врожденной мышечной дистрофии, Американская академия неврологии рекомендует пациентам контролировать сердечную функцию, дыхательные пути и желудочно-кишечный. Кроме того, считается, что речевая терапия, ортопедический и физические области, улучшили бы качество жизни человека.[3]
Хотя в настоящее время нет лекарств, важно сохранить мышечную активность и любую доступную коррекцию скелетных аномалий (например, сколиоза). Ортопедические процедуры, например спондилодез, поддерживает / увеличивает перспективу физического движения человека.[3]
Смотрите также
Рекомендации
- ^ а б c d е ж грамм час я j k Спаркс, Сьюзен; Quijano-Roy, Susana; Харпер, Эми; Рутковски, Энн; Гордон, Эринн; Хоффман, Эрик П .; Пегораро, Елена (01.01.1993). Пагон, Роберта А .; Адам, Маргарет П .; Ardinger, Holly H .; Уоллес, Стефани Э .; Амемия, Энн; Бин, Лора JH; Bird, Thomas D .; Фонг, Чин-То; Меффорд, Хизер С. (ред.). Обзор врожденной мышечной дистрофии. Сиэтл (Вашингтон): Вашингтонский университет, Сиэтл. PMID 20301468.обновление 2012
- ^ а б c «Обследование врожденной мышечной дистрофии: лабораторные исследования, визуализирующие исследования, другие тесты». emedicine.medscape.com. Получено 2016-04-28.
- ^ а б c «Врожденная мышечная дистрофия». Рекомендации Американской академии неврологии. 2015. Получено 28 апреля 2016.
- ^ Бертини, Энрико; Д'Амико, Адель; Гуаланди, Франческа; Петрини, Стефания (01.12.2011). «Врожденные мышечные дистрофии: краткий обзор». Семинары по детской неврологии. 18 (4): 277–288. Дои:10.1016 / j.spen.2011.10.010. ISSN 1071-9091. ЧВК 3332154. PMID 22172424.
- ^ «Гипотония: Медицинская энциклопедия MedlinePlus». www.nlm.nih.gov. Получено 2016-04-28.
- ^ Рид, Умбертина Конти (2009). «Врожденная мышечная дистрофия. Часть I: обзор фенотипических и диагностических аспектов». Arquivos de Neuro-Psiquiatria. 67 (1): 144–168. Дои:10.1590 / S0004-282X2009000100038. ISSN 0004-282X. PMID 19330236.
- ^ Рид, Умбертина (2009). «Врожденная мышечная дистрофия 2 часть» (PDF). Нейропсиквитрия. Получено 28 апреля 2016.
- ^ Мартин, Пол Т (2006). «Механизмы заболевания: врожденные мышечные дистрофии - гликозилирование занимает центральное место». Природа Клиническая Практика Неврология. 2 (4): 222–230. Дои:10.1038 / ncpneuro0155. ISSN 1745-834X. ЧВК 2855642. PMID 16932553.
- ^ а б c "Запись OMIM - № 253280 - МЫШЕЧНАЯ ДИСТРОФИЯ-ДИСТРОГЛИКАНОПАТИЯ (ВРОЖДЕННАЯ С АНОМАЛИЯМИ МОЗГА И ГЛАЗА), ТИП A, 3; MDDGA3". www.omim.org. Получено 2016-04-26.
- ^ «Ошибка 403».
- ^ Справка, Дом генетики. «Синдром Уокера-Варбурга». Домашний справочник по генетике. Получено 2016-04-26.
- ^ Quijano-Roy, Susana; Спаркс, Сьюзен; Рутковски, Энн (01.01.1993). Пагон, Роберта А .; Адам, Маргарет П .; Ardinger, Holly H .; Уоллес, Стефани Э .; Амемия, Энн; Бин, Лора JH; Bird, Thomas D .; Фонг, Чин-То; Меффорд, Хизер С. (ред.). Мышечная дистрофия, связанная с LAMA2. Сиэтл (Вашингтон): Вашингтонский университет, Сиэтл. PMID 22675738.обновление 2012
- ^ «Запись OMIM - № 612937 - ВРОЖДЕННОЕ ЗАБОЛЕВАНИЕ ГЛИКОЗИЛИРОВКИ, ТИП Io; CDG1O». www.omim.org. Получено 2016-04-26.
- ^ «Запись OMIM - № 615042 - ВРОЖДЕННОЕ ЗАБОЛЕВАНИЕ ГЛИКОЗИЛИРОВАНИЯ, ТИП Iu; CDG1U». www.omim.org. Получено 2016-04-26.
- ^ «Запись OMIM - № 608799 - ВРОЖДЕННОЕ ЗАБОЛЕВАНИЕ ГЛИКОЗИЛИРОВАНИЯ, ТИП Ie; CDG1E». www.omim.org. Получено 2016-04-26.
- ^ «Запись OMIM - № 602771 - МЫШЕЧНАЯ ДИСТРОФИЯ ЖЕСТКОГО ПОЗВОНОЧНИКА 1; RSMD1». www.omim.org. Получено 2016-04-26.
- ^ «Запись OMIM - № 613205 - МЫШЕЧНАЯ ДИСТРОФИЯ, ВРОЖДЕННАЯ, СВЯЗАННАЯ С LMNA». www.omim.org. Получено 2016-04-26.
- ^ "Запись OMIM - № 613204 - МЫШЕЧНАЯ ДИСТРОФИЯ, ВРОЖДЕННАЯ, ИЗ-ЗА ИНТЕГРИРОВАННОГО ДЕФИЦИТА ALPHA-7". www.omim.org. Получено 2016-04-26.
- ^ Справка, Дом генетики. «Врожденная мышечная дистрофия Фукуямы». Домашний справочник по генетике. Получено 2016-04-26.
- ^ "OMIM Entry - # 607855 - МЫШЕЧНАЯ ДИСТРОФИЯ, ВРОЖДЕННЫЙ МЕРОЗИН-ДЕФИЦИЕНТ, 1A; MDC1A". www.omim.org. Получено 2016-04-26.
- ^ "OMIM Entry -% 609456 - МЫШЕЧНАЯ ДИСТРОФИЯ, ВРОЖДЕННАЯ, МЕРОЗИНПОЗИТИВНАЯ". www.omim.org. Получено 2016-04-26.
- ^ "Запись OMIM - № 254090 - ВРОЖДЕННАЯ МЫШЕЧНАЯ ДИСТРОФИЯ УЛЛРИЧА 1; UCMD1". omim.org. Получено 2016-04-26.
- ^ Bönnemann, Carsten G .; Wang, Ching H .; Quijano-Roy, Susana; Деконинк, Николас; Бертини, Энрико; Феррейро, Ана; Мунтони, Франческо; Сьюри, Кэролайн; Беруд, Кристоф; Мэтьюз, Кэтрин Д.; Мур, Стивен А .; Беллини, Джонатан; Рутковски, Энн; Норт, Кэтрин Н. (1 апреля 2014 г.). «Диагностический подход к врожденным мышечным дистрофиям». Нервно-мышечные расстройства. 24 (4): 289–311. Дои:10.1016 / j.nmd.2013.12.011. ЧВК 5258110. PMID 24581957.
дальнейшее чтение
- А, Грациано; F, Бьянко; А, Д'Амико; Я, Мороний; S, Мессина; C, Бруно; E, Пегораро; М, Мора; Дж., Астрея (01.03.2015). «Распространенность врожденной мышечной дистрофии в Италии: исследование населения». Неврология. 84 (9): 904–911. Дои:10.1212 / WNL.0000000000001303. ISSN 0028-3878. ЧВК 4351663. PMID 25653289.
- Пако, Соня; Кассеррас, Тереза; Родригес, Мария Энджелс; Джоу, Кристина; Пучделлосес, Монтсеррат; Ортез, Карлос I .; Диас-Манера, Хорди; Галлардо, Эдуардо; Коломер, Жауме (15 декабря 2015). «Транскриптомный анализ фибробластов с врожденной мышечной дистрофией Ульриха выявляет сигнатуру внеклеточного матрикса заболевания и ключевые молекулярные регуляторы». PLOS One. 10 (12): e0145107. Bibcode:2015PLoSO..1045107P. Дои:10.1371 / journal.pone.0145107. ISSN 1932-6203. ЧВК 4686057. PMID 26670220.
- Фальсаперла, Рафаэле; Praticò, Andrea D .; Руджери, Мартино; Парано, Энрико; Риццо, Рената; Корселло, Джованни; Виталити, Джованна; Павоне, Пьеро (31 августа 2016 г.). «Врожденная мышечная дистрофия: от мышцы к мозгу». Итальянский педиатрический журнал. 42 (1): 78. Дои:10.1186 / s13052-016-0289-9. ISSN 1824-7288. ЧВК 5006267. PMID 27576556.
- «Резюме научно обоснованного руководства для ПАЦИЕНТОВ и их СЕМЬЕЙ ВРОЖДЕННАЯ МЫШЕЧНАЯ ДИСТРОФИЯ». aaan.com. Американская академия неврологии (AAN). Получено 5 декабря 2017.
внешняя ссылка
| Классификация | |
|---|---|
| Внешние ресурсы |
| Scholia имеет тема профиль для Врожденная мышечная дистрофия. |
| Типы | |
|---|---|
| Национальные / международные организации |
|
| Национальные / международные мероприятия |
|
| Клинические испытания | |