Синдром Робиноу - Robinow syndrome
| Синдром Робиноу | |
|---|---|
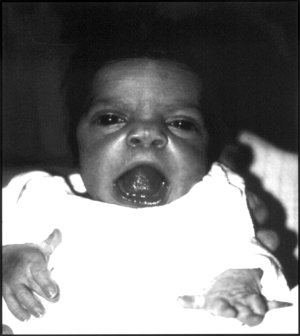 | |
| Младенец с чертами лица синдрома Робиноу. | |
| Специальность | Медицинская генетика |
Синдром Робиноу чрезвычайно редкий генетическое расстройство характеризуется короткими конечностями карликовость, патологии головы, лица и внешних гениталии, а также позвоночный сегментация. Заболевание было впервые описано в 1969 году человеком. генетик Мейнхард Робинов,[1] вместе с врачи Фредерик Н. Сильверман и Хьюго Д. Смит, в Американский журнал болезней детей. К 2002 году более 100 случаев были задокументированы и представлены в медицинской литературе.[1]
Существуют две формы расстройства: доминирующий и рецессивный, из которых первое более распространено. Пациенты с доминирующей версией часто умеренно страдают от вышеупомянутых симптомов. С другой стороны, рецессивные случаи обычно более выражены физически, и у людей может быть больше скелетный аномалии.[2] Рецессивная форма особенно часто встречается у индюк.[3] Однако, вероятно, это можно объяснить общий предок, поскольку семьи этих пациентов можно найти в одном городе на востоке Турции.[4] Кластеры аутосомный рецессивная форма также была зарегистрирована в Оман и Чехословакия.[1]
Синдром также известен как синдром Робиноу-Сильвермана-Смита, карликовость Робиноу, лицо плода, синдром лица плода,[5] синдром фации плода, акральный дизостоз с аномалиями лица и гениталий или синдром мезомелической карликовости и малых гениталий.[6] Рецессивная форма ранее была известна как синдром Ковесдема.
Признаки и симптомы


Робиноу отметил сходство лиц пораженных пациентов с лицами больного. плод, используя термин «фатальные фаски» для описания маленького лица и широко расставленных глаз.[1] Клинические признаки также могут включать короткий вздернутый нос, выступающий лоб и плоскую переносицу. Верхняя губа может быть «натянута»,[1] обнажая скученность зубов "галстук ", или же камедь гипертрофия.
Хотя глаза не выпячиваются, аномалии в нижних веко может произвести такое впечатление. Если глаза не могут полностью закрыться, может потребоваться операция. В дополнение уши может быть поставлен низко на голове или иметь деформированный ушная раковина.[нужна цитата ]
Пациенты страдают карликовостью, короткие предплечья, маленькие ступни и маленькие руки. Пальцы рук и ног также могут быть ненормально короткий и согнуты латерально или медиально. Большой палец может быть смещен, и некоторые пациенты, особенно в Турции, испытывают эктродактилия.[1] Все пациенты часто страдают аномалиями сегментации позвонков. Те, у кого преобладает вариант, имеют не более одного позвонок бабочки.[2] Однако пациенты с рецессивной формой могут страдать от полупозвонки, сращение позвонков и аномалии ребер. Некоторые случаи напоминают Синдром Ярхо-Левина или же спондилокостальный дизостоз.[нужна цитата ]
Генитальные дефекты, характерные для мужчин, включают: микропенис с нормально развитым мошонка и яички. Иногда яички могут не опускаться, или пациент может страдать от гипоспадия.[2] Пороки женских половых органов могут включать уменьшенный размер. клитор и слаборазвитые малые половые губы. Нечасто большие половые губы также может быть недоразвитым.[2] Некоторые исследования показали, что женщины могут испытывать атрезия влагалища или же гематоколпоз.[3]
Аутосомно-рецессивная форма заболевания, как правило, намного тяжелее. Примеры различий приведены в следующей таблице:[7]
| Характеристика | Аутосомно-рецессивный | Аутосомно-доминантный |
|---|---|---|
| Рост | Более низкий рост -2 SD или менее | Короткий или нормальный |
| Руки | Очень короткий | Немного короткий |
| Локоть | Вывих головки лучевой кости | Нет вывиха головки лучевой кости |
| Верхняя губа | Подогнутая верхняя губа | Нормальная верхняя губа |
| Смертность | 10% смертность | Без лишней смертности |
Сопутствующие условия
Медицинские условия включают частые инфекции уха, потеря слуха, гипотония, проблемы развития, проблемы с дыханием, трудности с питанием, светочувствительность, и пищеводный рефлюкс.[2]
Данные о плодородие и развитие вторичные половые признаки относительно редко. Сообщалось, что у пациентов мужского и женского пола были дети. Все репродуктивные самцы имели аутосомно-доминантную форму заболевания; фертильность пациентов с рецессивным вариантом неизвестна.[1]
Исследователи также сообщили об аномалиях в почечный тракт пораженных пациентов. Гидронефроз является относительно распространенным состоянием, и исследователи предположили, что это может привести к инфекция мочеиспускательного канала.[8] Кроме того, ряд пациентов страдали от кистозная дисплазия из почка.[1]
С синдромом Робиноу часто связывают ряд других состояний. Около 15% зарегистрированных пациентов страдают от врожденные пороки сердца. Хотя четкой закономерности нет, наиболее распространенные состояния включают: легочный стеноз и атрезия.[9] Кроме того, хотя интеллект в целом нормальный, около 15% пациентов обнаруживают задержку в развитии.[1]
Генетика
Генетический исследования связали аутосомно-рецессивную форму заболевания с ROR2 ген на позиции 9 длинной руки хромосома 9.[1] Ген отвечает за рост костей и хрящей. Этот же ген участвует в возникновении аутосомно-доминантного брахидактилия B.[1]

Аутосомно-доминантная форма связана с тремя генами - WNT5A, Сегмент полярности белка растрепанный гомолог DVL-1 (DVL1 ) и Сегмент полярности белка растрепанного гомолога DVL-3 (DVL3 ). Эта форма часто вызывается новыми мутациями и обычно менее серьезна, чем рецессивная форма. Еще два гена были связаны с этим заболеванием - Frizzled-2 (FZD2 ) и Нуклеоредоксин (Ген NXN ).[10] Все эти гены принадлежат к одному метаболическому пути - системе WNT. Эта система участвует в секреции различных соединений как у плода, так и у взрослого человека.[нужна цитата ]
Плод УЗИ может предложить пренатальная диагностика 19 недель в беременность. Однако характеристики плода, страдающего более легкой доминантной формой, не всегда легко отличить от более серьезного рецессивного случая. Генетическое консультирование - вариант при наличии семейного анамнеза.[1]
Диагностика
Синдром Робиноу подозревается на основании клинических данных и семейного анамнеза и подтверждается типичными двуаллельными патогенными вариантами ROR-2, идентифицированными с помощью молекулярно-генетического тестирования.[11]
Уход
Лечением различных проявлений обычно занимается многопрофильная бригада.[12]
История
Заболевание было впервые описано в 1969 г. Немецко-американским исследованием людей. Генетик Мейнхард Робинов (1909–1997),[1] вместе с врачами Фредериком Н. Сильверманом и Хьюго Д. Смитом в Американский журнал болезней детей. К 2002 году более 100 случаев были задокументированы и представлены в медицинской литературе.[1]
Рекомендации
- ^ а б c d е ж грамм час я j k л м п Паттон, Массачусетс; Афзал А. Р. (2002). «Синдром Робинова». Журнал медицинской генетики. 39 (5): 305–10. Дои:10.1136 / jmg.39.5.305. ЧВК 1735132. PMID 12011143.
- ^ а б c d е Фонд синдрома Робинова. Общая информация. По состоянию на 19 мая 2006 г.
- ^ а б Бальчи, Севим; Бексач, Синан; Халилоглу, Митхат; Эрджис, Мурат; Эриилмаз, Музаффер (1998). «Синдром Робинова, атрезия влагалища, гематоколпоз и лишний средний палец». Американский журнал медицинской генетики. 79 (1): 27–9. Дои:10.1002 / (SICI) 1096-8628 (19980827) 79: 1 <27 :: AID-AJMG7> 3.0.CO; 2-F. PMID 9738864.
- ^ Бруннер, Хан Г; Ван Боховен, Ганс; Челли, Якопо; Кайсерили, Хюля; Ван Бойзеком, Эллен; Бальчи, Севим; Брюссель, Вим; Сковби, Флемминг; Керр, Бронвин; Percin, E. Ferda; Акарсу, Нуртен (2000). «Мутация гена, кодирующего тирозинкиназу ROR2, вызывает аутосомно-рецессивный синдром Робиноу». Природа Генетика. 25 (4): 423–6. Дои:10.1038/78113. PMID 10932187.
- ^ Национальная организация редких заболеваний, Inc. Синдром Робинова. Последнее изменение 15 мая 2006 г. Проверено 19 мая 2006 г.
- ^ База данных синдромов Яблонского. Синдромы множественной врожденной аномалии / умственной отсталости (MCA / MR). Доступ 20 мая 2006 г.
- ^ Робиноу, М. (1993). «Синдром Робиноу (лицо плода)». Клиническая дисморфология. 2 (3): 189–98. Дои:10.1097/00019605-199307000-00001. PMID 8287180.
- ^ Шпринцен, Роберт Дж; Goldberg, R.B; Saenger, P; Сидоти, Э. Дж. (1982). «Передача синдрома Робиноу от мужчины к мужчине». Американский журнал болезней детей. 136 (7): 594–7. Дои:10.1001 / архпеди.1982.03970430026007. PMID 7091086.
- ^ Уэббер, Стивен А; Варговски, Дэвид С; Читаят, Давид; Сандор, Джордж Г. С (1990). «Врожденный порок сердца и синдром Робинова: совпадение или дополнительный компонент синдрома?». Американский журнал медицинской генетики. 37 (4): 519–21. Дои:10.1002 / ajmg.1320370418. PMID 2260599.
- ^ Уайт, Янсон Дж; Маццеу, Юлиана Ф .; Кобан-Акдемир, Зейнеп; Байрам, Явуз; Бахрамбейги, Вахид; Хойшен, Александр; Ван Бон, Брегье В.М.; Гездиричи, Альпер; Гулек, Элиф Йилмаз; Рамонд, Фрэнсис; Турень, Рено; Тевенон, Жюльен; Шинави, Марван; Бивер, Эрин; Хили, Дженнифер; Гувер-Фонг, Джули; Дурмаз, Серен Д; Карабулут, Халил Гурхан; Марзиоглу-Оздемир, Эбру; Каир, Атилла; Duz, Mehmet B; Семь, Мехмет; Прайс, Сьюзан; Феррейра, Барбара Мерфорт; Вианна-Морганте, Анджела М; Эллард, Сиан; Пэрриш, Эндрю; Стальс, Карен; Флорес-Дабуб, Хосуэ; и другие. (2018). «Нарушения передачи сигналов WNT лежат в основе генетической гетерогенности синдрома Робиноу». Американский журнал генетики человека. 102 (1): 27–43. Дои:10.1016 / j.ajhg.2017.10.002. ЧВК 5777383. PMID 29276006.
- ^ Афзал А.Р., Джеффри С. (июль 2003 г.). «Один ген, два фенотипа: мутации ROR2 при аутосомно-рецессивном синдроме Робиноу и аутосомно-доминантной брахидактилии типа B». Гм. Мутат. 22 (1): 1–11. Дои:10.1002 / humu.10233. PMID 12815588.
- ^ Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, Roifman M, Brunner H, Lohr J, Mazzeu J, Chitayat D (октябрь 2019 г.). Аутосомно-доминантный синдром Робиноу в GeneReviews. PMID 25577943.
дальнейшее чтение
- Уайт, Янсон; Маццеу, Юлиана Ф .; Хойшен, Александр; Джангиани, Шалини Н; Гамбин, Томаш; Альчино, Микеле Калижорн; Пенни, Саманта; Сараива, Хорхе М; Хов, Ханне; Сковби, Флемминг; Кайсерили, Хюля; Эстрелла, Элисия; Вулто-Ван Силфхаут, Аннеке Т; Стихауэр, Марлоуз; Музны, Донна М; Саттон, В. Рид; Гиббс, Ричард А; Лупски, Джеймс Р.; Бруннер, Хан Г; Ван Бон, Брегье В.М.; Карвалью, Клаудиа МБ (2015). "Кластеризация мутаций сдвига кадра DVL1 в предпоследнем экзоне вызывает аутосомно-доминантный синдром Робинова". Американский журнал генетики человека. 96 (4): 612–22. Дои:10.1016 / j.ajhg.2015.02.015. ЧВК 4385180. PMID 25817016.
внешняя ссылка
| Классификация | |
|---|---|
| Внешние ресурсы |