Синдром Нунана - Noonan syndrome
| Синдром Нунана | |
|---|---|
| Другие имена | Мужской синдром Тернера, синдром Нунана-Эмке, синдром Тернера, синдром Ульриха-Нунана[1] |
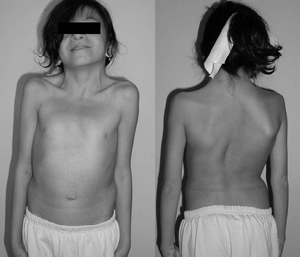 | |
| 12-летняя девочка с синдромом Нунана. Типичный перепончатая шея. Двойная структурная кривая с реберной деформацией. | |
| Специальность | Медицинская генетика, педиатрия |
| Симптомы | Слегка необычные черты лица, небольшой рост, врожденный порок сердца, проблемы с кровотечением, пороки развития скелета[1] |
| Осложнения | Лейкемия[1] |
| Обычное начало | Присутствует при рождении[2] |
| Типы | Типы с 1 по 6[3] |
| Причины | Генетическая мутация (аутосомно-доминантный )[1] |
| Диагностический метод | Предполагается на основании симптомов, подтверждено генетическое тестирование[4][2] |
| Дифференциальная диагностика | Кардиофациально-кожный синдром, Синдром Тернера, Синдром Костелло, нейрофиброматоз 1 типа[2][3] |
| Уход | По симптомам[3] |
| Медикамент | Гормон роста[3] |
| Прогноз | Зависит от тяжести проблем с сердцем[3] |
| Частота | 1 из 100 (1 из 2000 тяжелых заболеваний)[4] |
Синдром Нунана (NS) это генетическое расстройство которые могут проявляться слегка необычными чертами лица, небольшим ростом, врожденный порок сердца, проблемы с кровотечением, и пороки развития скелета.[1] Особенности лица включают: широко расставленные глаза, светлые глаза, низко посаженные уши, короткая шея и маленькая нижняя челюсть.[1] Проблемы с сердцем могут включать стеноз клапана легочной артерии.[1] В Грудина может либо выступать или быть затонувший, в то время как позвоночник может быть аномально изогнут.[1] Интеллект при синдроме часто нормальный.[1] Осложнения NS могут включать: лейкемия.[1]
Номер генетические мутации может привести к синдрому Нунана.[1] Состояние может быть унаследовано от родителей человека в качестве аутосомно-доминантный состояние или возникать как новая мутация.[3][1] Синдром Нунана - это разновидность РАСопатия, лежащий в основе механизма, который включает чрезмерную активацию внутри RAS / MAPK клеточная сигнализация путь.[1] Диагноз можно заподозрить на основании симптомов, медицинская визуализация, и анализы крови.[2][4] Подтверждение может быть достигнуто с помощью генетическое тестирование.[2]
Лекарства от NS не известно.[5] Лечение основано на симптомах и основных проблемах, и может потребоваться дополнительная поддержка в школе.[3] Гормональная терапия роста в детстве может увеличить окончательный рост пострадавшего.[3] Долгосрочные результаты обычно зависят от серьезности сердечных заболеваний.[3]
По оценкам, 1 из 100 человек страдает от NS в легкой форме, в то время как примерно у 1 из 2000 есть более тяжелая форма заболевания.[4] Мужчины поражаются чаще, чем женщины.[2] Состояние было впервые описано в 1883 году и названо в честь американского педиатрический кардиолог Жаклин Нунан, который описал дальнейшие случаи в 1963 году.[2]
Признаки и симптомы


Наиболее распространенные признаки, ведущие к диагностике синдрома Нунан являются уникальными характеристиками лица и опорно-двигательного аппарата особенности. Черты лица наиболее заметны в младенчестве и становятся менее очевидными с возрастом у многих людей с синдромом Нунана.[7]
Голова
Некоторые из характерных черт синдрома Нунана включают: большая голова с избытком кожи на задней части шеи, низкой линией роста волос на затылке, высокой линией роста волос на передней части головы, треугольной формой лица, широким лбом и короткой перепончатой шеей.[нужна цитата ]
В глазах, гипертелоризм (широко посаженные глаза) - определяющая характеристика, которая присутствует у 95% людей с синдромом Нунана. Это может сопровождаться эпикантальные складки (лишняя кожная складка у внутреннего уголка глаза), птоз (опущение век), проптоз (выпученные глаза), косоглазие (поворот глаз внутрь или наружу), нистагм (подергивание глаз) и рефракционные нарушения зрения.
Нос может быть маленьким, широким и вздернутым.
У людей с синдромом Нунана может быть нарушено развитие ушей и слуховой системы. Это может привести к низко посаженным ушам (более 90%), обратному вращению ушей (более 90%), толстой спирали (внешний край) уха (более 90%), неполному складыванию ушей, хроническому средний отит (ушные инфекции) и потеря слуха.
При синдроме Нунана также может быть нарушено развитие ротовой полости. Это может привести к образованию глубоких бороздок желобка (линии верхней губы) (более 90%), микрогнатия (заниженная нижняя челюсть), высокое сводчатое небо, трудности с артикуляцией (зубы не совпадают), что может привести к проблемам с зубами. Подобно мышечным проявлениям, описанным выше, во рту может наблюдаться плохой контроль языка.
Кожа
Кожные признаки и симптомы синдрома Нунана включают: лимфедема (лимфатический отек конечностей), келоид образование, чрезмерное образование рубцов, гиперкератоз (чрезмерное развитие внешнего кожного слоя), пигментные невусы (темные пигментные пятна на коже), и заболевание соединительной ткани
Опорно-двигательный
При синдроме Нунана могут возникать аномалии конечностей и конечностей. Это может проявляться в виде тупых кончиков пальцев, лишних подушечек на пальцах рук и ног, отек тыльной стороны кистей рук и верхней части стоп, а также Cubitus valgus (широкий угол наклона локтей).
При низком росте гормон роста иногда комбинируют с IGF-1 (или в качестве альтернативы IGF-1 как автономный) можно использовать для более быстрого достижения увеличенного / конечного роста. Окончательный рост взрослого человека с синдромом Нунана составляет около 161–167 см у мужчин и 150–155 см у женщин, что приближается к нижней границе нормы.[8]
Спинальные аномалии могут присутствовать до 30% времени, и более чем в 60% этих случаев может потребоваться хирургическое вмешательство.[нужна цитата ] Другие скелетно-мышечные проявления при синдроме Нунана связаны с недифференцированными нарушениями соединительной ткани, которые могут быть связаны с контрактурами суставов (стянутостью) или гипермобильностью суставов (рыхлостью). Дополнительные факторы могут проявляться в виде выпуклости лопатки, сколиоза, выпуклости грудной кости (pectus carinatum), вдавления грудной кости (pectus excatum). Мышечные аномалии могут проявляться в виде гипотонии (низкого мышечного тонуса), что может привести к лордозу (усиление впадины в спине) из-за плохого мышечного тонуса живота.
Сердце
Синдром Нунана - вторая по частоте синдромная причина врожденных пороков сердца. Это включает стеноз клапана легочного клапана (50–60%), дефекты межпредсердной перегородки (10–25%), желудочковый дефекты перегородки (5–20%) и гипертрофическая кардиомиопатия (12–35%).[нужна цитата ]
Легкие
У некоторых людей сообщалось об ограничительной функции легких.
Желудочно-кишечный тракт
С синдромом Нунана связан ряд разнообразных желудочно-кишечных (ЖКТ) симптомов. К ним относятся трудности с глотанием, низкая перистальтика кишечника, гастропарез (задержка опорожнения желудка), кишечная мальротация, и частые или решительные рвота. Эти проблемы с пищеварением могут привести к снижение аппетита, неспособность процветать от младенчества до половой зрелости (75%), а иногда и потребность в зонде для кормления.
Мочеполовой системы
У некоторых мужчин с синдромом Нунана яички не опускаются (крипторхизм ).
Тираж
Лимфатические аномалии, включая задние шейные гигрома (перепончатая шея) и Лимфедема может присутствовать у людей с синдромом Нунана.
С синдромом Нунана связан ряд нарушений свертываемости крови, в том числе дисфункция тромбоцитов, Свертывание крови расстройства, частичная недостаточность фактор VIII: C, частичный дефицит фактор XI: C, частичный дефицит фактор XII: C и дисбаланс активности ингибитора активатора плазминогена типа 1 (PAI-1) и тканевого активатора плазминогена (t-PA).[нужна цитата ] Это было связано с Болезнь фон Виллебранда, Амегакариоцитарная тромбоцитопения (низкое количество тромбоцитов), длительное действие частичное тромбопластиновое время, в сочетании дефекты коагуляции. При наличии этих сопутствующих расстройств, связанных с синдромом Нунана, они могут быть связаны с предрасположенностью к появлению синяков или кровотечений.
Неврологический
Изредка, Мальформация Киари (тип 1), что может привести к гидроцефалия.[нужна цитата ] Сообщалось также об изъятиях.
Причины

Рецидив у братьев и сестер и очевидная передача от родителя к ребенку уже давно указывают на генетический дефект с аутосомно-доминантный наследование и выражение переменной. Мутации в Рас /митоген-активированная протеинкиназа сигнальные пути, как известно, ответственны за около 70% случаев NS.[9]
Люди с NS имеют до 50% шансов передать его своим потомкам. Тот факт, что пораженный родитель не всегда идентифицируется для детей с NS, предполагает несколько возможностей:
- Проявления могут быть настолько незаметными, что их нельзя распознать (переменная выразительность )
- NS является неоднородный, включающий более одного аналогичного состояния по разным причинам, и некоторые из них могут не передаваться по наследству.
- Большая часть случаев может представлять собой новые, спорадические мутации.
| Тип | Онлайн Менделирующее наследование в базе данных Man | Ген | Год найден | Locus | % случаев | Описание | Ссылка |
|---|---|---|---|---|---|---|---|
| NS1 | 163950 | ПТПН11 | 2001 | 12q24.1 | 50% | В ПТПН11 ген кодирует протеинтирозинфосфатаза ШП-2. Этот белок входит в состав нескольких внутриклеточных преобразование сигнала пути, участвующие в эмбриональном развитии, которые модулируют клеточное деление, дифференцировку и миграцию, в том числе один, опосредованный фактор роста эпидермиса рецептор, что важно при формировании полулунной сердечные клапаны. Дупликация участка хромосомы, содержащего ПТПН11 также может привести к NS. | [10] [11] |
| NS2 | 605275 | Неизвестный; аутосомно-рецессивный | [12] | ||||
| NS3 | 609942 | KRAS | 2006 | 12п12.1 | <5% | [13] | |
| NS4 | 610733 | SOS1 | 2006 | 2п21 | 10% | Активация мутаций в SOS1 может дать начало NS. ШП-2 и SOS1 положительно регулировать Рас /MAP киназа путь, предполагая, что его нарушение регуляции опосредует развитие NS.[14] | [15] |
| NS5 | 611553 | RAF1 | 2007 | 3п25 | 3–17% | [16] |
Гетерозиготные мутации в NRAS, HRAS, BRAF, SHOC2, MAP2K1, MAP2K2, и CBL также были связаны с меньшим процентом NS и родственных фенотипов.[17]
Состояние, известное как "нейрофиброматоз-синдром Нунана " связан с нейрофибромин.[18]
Диагностика
NS может быть подтверждено генетически по наличию любой из известных мутаций, перечисленных выше. Однако, несмотря на идентификацию 14 причинных генов, отсутствие известной мутации не исключает диагноз, так как большее количество еще не открытых генов могут вызывать NS. Таким образом, диагноз НС по-прежнему основывается на клинических особенностях. Другими словами, это делается, когда врач чувствует, что у человека достаточно функций, чтобы оправдать этикетку. Основные ценности генетического диагноза заключаются в том, что он направляет дополнительные медицинские оценки и оценки развития, исключает другие возможные объяснения особенностей и позволяет более точно оценить риск рецидива. При проведении большего числа исследований корреляции генотип-фенотип положительный генетический диагноз поможет клиницисту узнать о возможных аномалиях, специфичных для данной мутации определенного гена. Например, учащение гипертрофической кардиомиопатии наблюдается у людей с мутацией KRAS и повышенный риск ювенильного миеломоноцитарного лейкоза существует из-за мутации ПТПН11. В будущем исследования могут привести к целенаправленному лечению симптомов NS, которое зависит от того, какая генетическая мутация есть у человека.
До рождения
Пренатальные особенности, которые могут побудить врачей рассмотреть возможность диагноза синдрома Нунана, включают кистозную гигрому, повышенную прозрачность затылочной кости, плевральный выпот и отек.[19]
Дифференциальная диагностика
Пока Синдром Тернера имеет сходство с почечными аномалиями и задержкой развития, синдром Тернера встречается только у женщин и часто проявляется по-разному. При синдроме Тернера наблюдается меньшая частота задержки развития, левосторонние пороки сердца являются постоянными, а возникновение почечных аномалий намного ниже.[20]
Другие РАСопатии
- Синдром Ватсона - Синдром Ватсона имеет ряд сходных характеристик с синдромом Нунана, таких как низкий рост, стеноз легочного клапана, переменное интеллектуальное развитие и изменения пигмента кожи.[20][21]
- Кардиофациально-кожный (CFC) синдром - CFC-синдром очень похож на синдром Нунана из-за схожих сердечных и лимфатических функций. Однако при синдроме CFC умственная отсталость и желудочно-кишечные проблемы часто бывают более серьезными и выраженными.[20][22]
- Синдром Костелло - Как и синдром CFC, синдром Костелло имеет общие черты с синдромом Нунана. Однако состояния можно различить по их генетической причине.[23][24]
- Нейрофиброматоз 1 (NF1) [20][25]
- Синдром Вильямса [20][26]
Управление
Лечение варьируется в зависимости от осложнений, но, как правило, является стандартным, отражающим лечение населения в целом.[20] Руководства по лечению, разделенные по системам, включая общие, связанные с развитием, стоматологические, рост и питание, сердечно-сосудистые, аудиологические, гематологические, почечные и скелетные, которые учитывают действия, которые необходимо предпринять при постановке диагноза, после постановки диагноза и в случае появления симптомов, были опубликованы американцем консорциум.[19]
В частности, лечение сердечно-сосудистых осложнений аналогично лечению у населения в целом, а лечение кровоточащего диатеза основывается на дефиците конкретного фактора или агрегации тромбоцитов.[20]
- Нейропсихологическое тестирование рекомендуется для выявления сильных сторон и проблем, необходимых для поддержки, необходимой для учебы и карьеры.
- Образовательная настройка, такая как индивидуальная программа обучения план иногда требуется для детей школьного возраста.
- Логопедия при наличии проблем с речью и артикуляцией
- Физическая терапия и трудотерапия при задержке крупной и мелкой моторики
- Гипотония и двигательные проблемы часто влияют на почерк. Приспособления для уменьшения требований к почерку улучшат производительность и сохранят функцию рук в долгосрочной перспективе.
- Рекомендуется периодическое наблюдение и постоянный мониторинг нарушений, обнаруженных в любой системе, особенно сердечно-сосудистой.[27][8]
Риск анестезии
Хотя сообщалось, что у нескольких людей с синдромом Нунана развиваются злокачественная гипертермия мутация гена заболеваний, которые, как известно, связаны со злокачественной гипертермией, отличается от генной мутации синдрома Нунана.[28]
Прогноз
Продолжительность жизни людей с синдромом Нунана может быть аналогична продолжительности жизни населения в целом, однако синдром Нунана может быть связан с несколькими состояниями здоровья, которые могут способствовать смертности. Наибольший вклад в смертность людей с синдромом Нунана вносят осложнения сердечно-сосудистых заболеваний.[29][8] Таким образом, прогноз во многом зависит от наличия или отсутствия сердечного заболевания, а также от типа и степени тяжести заболевания (если болезнь присутствует).[8] В частности, синдром Нунана с гипертрофической кардиомиопатией связан с повышенной смертностью.[29][8]
История
Жаклин Нунан практиковал детским кардиологом в Университет Айовы когда она заметила, что дети с редким пороком сердца, клапанный легочный стеноз, часто имели характерный внешний вид, с невысокий рост, перепончатая шея, широко расставленные глаза, и низко посаженные уши. Пострадали и мальчики, и девочки. Эти характеристики иногда наблюдались в семьях, но не были связаны с хромосомный аномалии. Она изучила 833 человека с синдромом Нунана в клинике врожденных пороков сердца, ища другие врожденные аномалии, и в 1963 году представила доклад: «Ассоциированные внесердечные пороки развития у детей с врожденными пороками сердца».[30] Описано 9 детей, у которых помимо врожденного порока сердца были характерные черты лица, деформация грудной клетки и низкий рост.
Доктор Джон Опитц, бывший ученик доктора Нунана, впервые начал называть это состояние «синдромом Нунана», когда увидел детей, похожих на тех, кого описал доктор Нунан. В 1968 году доктор Нунан выпустила статью под названием «Гипертелоризм с фенотипом Тернера», в которой она изучила 19 пациентов, у которых проявлялись симптомы, указывающие на синдром Нунана.[31] В 1971 году на Симпозиуме сердечно-сосудистых дефектов название «синдром Нунана» было официально признано.
Рекомендации
- ^ а б c d е ж грамм час я j k л м «Синдром Нунана». Домашний справочник по генетике. Получено 24 декабря 2018.
- ^ а б c d е ж грамм «Синдром Нунана». NORD (Национальная организация по редким заболеваниям). 2016. Получено 24 декабря 2018.
- ^ а б c d е ж грамм час я «Синдром Нунана». Информационный центр по генетическим и редким заболеваниям (GARD) - программа NCATS. Получено 25 декабря 2018.
- ^ а б c d Bhambhani V, Muenke M (январь 2014 г.). «Синдром Нунана». Американский семейный врач. 89 (1): 37–43. ЧВК 4099190. PMID 24444506.
- ^ «Синдром Нунана - проблемы со здоровьем детей». Руководства Merck для потребителей. Получено 25 декабря 2018.
- ^ а б Носан Г., Берток С., Весел С., Интема Х. Г., Паро-Панджан Д. (декабрь 2013 г.). «Смертельное течение гипертрофической кардиомиопатии при синдроме Нунана из-за новой мутации зародышевой линии в гене KRAS: тематическое исследование». Хорватский медицинский журнал. 54 (6): 574–8. Дои:10.3325 / cmj.2013.54.574. ЧВК 3893993. PMID 24382853.
- ^ Романо, Алисия А .; Allanson, Judith E .; Дальгрен, Джованна; Gelb, Bruce D .; Холл, Брайан; Пьерпон, Мэри Элла; Робертс, Эми Э .; Робинсон, Ванда; Takemoto, Clifford M .; Нунан, Жаклин А. (октябрь 2010 г.). «Синдром Нунана: клинические особенности, диагностика и рекомендации по лечению». Педиатрия. 126 (4): 746–759. Дои:10.1542 / педс.2009-3207. ISSN 1098-4275. PMID 20876176. S2CID 11297756.
- ^ а б c d е Allanson, Judith E .; Робертс, Эми Э. (1993), Адам, Маргарет П .; Ardinger, Holly H .; Пагон, Роберта А .; Уоллес, Стефани Э. (ред.), «Синдром Нунана», GeneReviews®, Вашингтонский университет, Сиэтл, PMID 20301303, получено 2019-11-18
- ^ Раззак М.А., Комоике Й., Нисидзава Т., Инаи К., Фурутани М., Хигасинакагава Т., Мацуока Р. (март 2012 г.). «Характеристика новой мутации KRAS, выявленной при синдроме Нунан». Американский журнал медицинской генетики. Часть А. 158A (3): 524–32. Дои:10.1002 / ajmg.a.34419. PMID 22302539. S2CID 34135931.
- ^ Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, et al. (Декабрь 2001 г.). «Мутации в PTPN11, кодирующем протеинтирозинфосфатазу SHP-2, вызывают синдром Нунана». Природа Генетика. 29 (4): 465–8. Дои:10,1038 / ng772. PMID 11704759. S2CID 14627986.
- ^ Щелочков О.А., Патель А., Weissenberger GM, Chinault AC, Wiszniewska J, Fernandes PH и др. (Апрель 2008 г.). «Дублирование полосы хромосомы 12q24.11q24.23 приводит к явному синдрому Нунана». Американский журнал медицинской генетики. Часть А. 146A (8): 1042–8. Дои:10.1002 / ajmg.a.32215. PMID 18348260. S2CID 205309115.
- ^ ван дер Бургт I, Бруннер Х (сентябрь 2000 г.). «Генетическая гетерогенность в синдроме Нунана: доказательства аутосомно-рецессивной формы». Американский журнал медицинской генетики. 94 (1): 46–51. Дои:10.1002 / 1096-8628 (20000904) 94: 1 <46 :: AID-AJMG10> 3.0.CO; 2-I. PMID 10982482.
- ^ Шубберт С., Ценкер М., Роу С.Л., Бёлль С., Кляйн С., Боллаг Г. и др. (Март 2006 г.). «Мутации зародышевой линии KRAS вызывают синдром Нунана». Природа Генетика. 38 (3): 331–6. Дои:10,1038 / ng1748. PMID 16474405. S2CID 8193354.
- ^ Бентирес-Элдж М., Контаридис М.И., Нил Б.Г. (март 2006 г.). «Остановки на пути RAS при генетических заболеваниях человека». Природа Медицина. 12 (3): 283–5. Дои:10,1038 / нм0306-283. PMID 16520774. S2CID 6989331.
- ^ Робертс А.Е., Араки Т., Суонсон К.Д., Монтгомери К.Т., Скирипо Т.А., Джоши В.А. и др. (Январь 2007 г.). «Мутации увеличения функции зародышевой линии в SOS1 вызывают синдром Нунана». Природа Генетика. 39 (1): 70–4. Дои:10,1038 / ng1926. PMID 17143285. S2CID 10222262.
- ^ Razzaque MA, Nishizawa T., Komoike Y, Yagi H, Furutani M, Amo R и др. (Август 2007 г.). «Мутации увеличения функции зародышевой линии в RAF1 вызывают синдром Нунана». Природа Генетика. 39 (8): 1013–7. Дои:10,1038 / ng2078. PMID 17603482. S2CID 29753972.
- ^ "Скачать каталог - Медицинские лаборатории Мэйо". www.mayomedicallaboratories.com.
- ^ Де Лука А., Боттилло I, Саркози А., Карта С, Нери С., Беллаккио Э и др. (Декабрь 2005 г.). «Мутации гена NF1 представляют собой главное молекулярное событие, лежащее в основе синдрома нейрофиброматоза-Нунана». Американский журнал генетики человека. 77 (6): 1092–101. Дои:10.1086/498454. ЧВК 1285166. PMID 16380919.
- ^ а б Робертс, Эми Э .; Allanson, Judith E .; Тарталья, Марко; Гелб, Брюс Д. (26 января 2013 г.). «Синдром Нунана». Ланцет. 381 (9863): 333–342. Дои:10.1016 / S0140-6736 (12) 61023-X. ISSN 1474-547X. ЧВК 4267483. PMID 23312968.
- ^ а б c d е ж грамм Allanson, Judith E .; Робертс, Эми Э. (1993), Адам, Маргарет П .; Ardinger, Holly H .; Пагон, Роберта А .; Уоллес, Стефани Э. (ред.), «Синдром Нунана», GeneReviews®, Вашингтонский университет, Сиэтл, PMID 20301303, получено 2019-11-25
- ^ Allanson, J.E .; Упадхьяя, М .; Watson, G.H .; Partington, M .; MacKenzie, A .; Lahey, D .; MacLeod, H .; Сарфарази, М .; Broadhead, W .; Харпер, П. С. (ноябрь 1991 г.). «Синдром Ватсона: это подтип нейрофиброматоза 1 типа?». Журнал медицинской генетики. 28 (11): 752–756. Дои:10.1136 / jmg.28.11.752. ISSN 0022-2593. ЧВК 1017110. PMID 1770531.
- ^ Броня, C. M .; Аллансон, Дж. Э. (апрель 2008 г.). «Дальнейшее определение кардио-фациально-кожного синдрома: клинические особенности 38 человек с доказанными мутациями». Журнал медицинской генетики. 45 (4): 249–254. Дои:10.1136 / jmg.2007.054460. ISSN 1468-6244. PMID 18039946. S2CID 9742395.
- ^ Kerr, B .; Delrue, M.-A .; Sigaudy, S .; Perveen, R .; Marche, M .; Бургелин, И .; Стеф, М .; Тан, В .; Eden, O.B .; О'Салливан, Дж .; Де Сандре-Джованноли, А. (май 2006 г.). «Генотип-фенотипическая корреляция при синдроме Костелло: анализ мутаций HRAS в 43 случаях». Журнал медицинской генетики. 43 (5): 401–405. Дои:10.1136 / jmg.2005.040352. ISSN 1468-6244. ЧВК 2564514. PMID 16443854.
- ^ Грипп, Карен В .; Lin, Angela E .; Stabley, Deborah L .; Николсон, Линда; Скотт, Чарльз I; Дойл, Дэниел; Аоки, Йоко; Мацубара, Йоичи; Zackai, Elaine H .; Лапунзина, Пабло; Гонсалес-Менесес, Антонио (01.01.2006). «Анализ мутаций HRAS при синдроме Костелло: корреляция генотипа и фенотипа». Американский журнал медицинской генетики, часть A. 140A (1): 1–7. Дои:10.1002 / ajmg.a.31047. ISSN 1552-4825. PMID 16329078. S2CID 27334655.
- ^ Бертола, Дебора Р .; Pereira, Alexandre C .; Пассетти, Фабио; de Oliveira, Paulo S.L .; Мессиан, Людвин; Gelb, Bruce D .; Kim, Chong A .; Кригер, Хосе Эдуардо (30 июля 2005 г.). «Синдром нейрофиброматоза-Нунана: Молекулярные доказательства сочетания обоих расстройств у пациента». Американский журнал медицинской генетики, часть A. 136A (3): 242–245. Дои:10.1002 / ajmg.a.30813. ISSN 1552-4825. PMID 15948193. S2CID 40235559.
- ^ Моррис, Коллин А. (1993), Адам, Маргарет П .; Ardinger, Holly H .; Пагон, Роберта А .; Уоллес, Стефани Э. (ред.), «Синдром Вильямса», GeneReviews®, Вашингтонский университет, Сиэтл, PMID 20301427, получено 2019-11-25
- ^ «Синдром Нунан - Симптомы, диагностика и лечение | BMJ Best Practice». bestpractice.bmj.com. Получено 2019-11-18.
- ^ "Повышает ли синдром Нунана восприимчивость к злокачественной гипертермии? - MHAUS". www.mhaus.org. Получено 2019-11-25.
- ^ а б «ДинаМед». www.dynamed.com. Получено 2019-11-11.
- ^ Нунан Дж. А., Эмке Д. А. (1963). «Сопутствующие некардиальные пороки развития у детей с врожденными пороками сердца». Midwest Soc Pediatr Res. 63: 468–70.
- ^ Нунан, Жаклин А. (1968-10-01). «Гипертелоризм с фенотипом Тернера: новый синдром с ассоциированным врожденным пороком сердца». Американский журнал болезней детей. 116 (4): 373–80. Дои:10.1001 / архпеди.1968.02100020377005. ISSN 0002-922X. PMID 4386970.
внешняя ссылка
| Классификация | |
|---|---|
| Внешние ресурсы |