Синдром Смита – Файнмана – Майерса - Smith–Fineman–Myers syndrome
Эта статья нужны дополнительные цитаты для проверка. (Август 2010 г.) (Узнайте, как и когда удалить этот шаблон сообщения) |
| Синдром Смита – Файнмана – Майерса | |
|---|---|
| Другие имена | Х-сцепленная умственная отсталость-гипотонический фациальный синдром 1 (MRXHF1), Синдром Карпентера – Вазири, Синдром Чадли – Лоури, SFMS, Синдром Холмса-Банды и Синдром Юберга-Марсиди (JMS),[1][2] |
 | |
| Синдром Смита – Файнмана – Майерса имеет Х-сцепленный рецессивный образец наследования. | |
Синдром Смита – Файнмана – Майерса (SFMS1) - это врожденное заболевание что вызывает врожденные дефекты. Этот синдром был назван в честь Ричарда Д. Смита, Роберта М. Файнмана и Гарта Дж. Майерса, которые обнаружили его около 1980 года.[3]
Признаки и симптомы
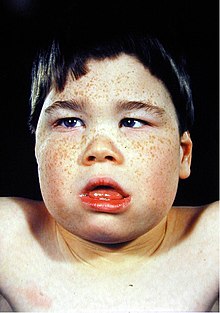
SFMS влияет на скелетную и нервную систему. Внешними признаками этого синдрома являются необычный внешний вид лица с немного меньшими размерами головы и необычной формы, узкое лицо, которое также называют долихоцефалия, большой рот с опущенной нижней губой, которая держится открытой, выступающая верхняя челюсть, широко расставленные передние верхние зубы, недоразвитый подбородок, волчья пасть и экзотропно раскосые глаза с опущенными веками.
Мужчины с SFMS имеют низкий рост и худощавое телосложение. Также кожа слегка пигментирована с множеством веснушек. Они могут иметь сколиоз и аномалии грудной клетки.
У больных мальчиков снижен мышечный тонус в младенчестве и раннем детстве. Рентгеновские лучи иногда показывают, что их кости недоразвиты и показывают характеристики более молодых костей детей. У мальчиков в возрасте до 10 лет обычно снижается мышечный тонус, но позже у пациентов с SFMS в возрасте старше 10 лет наблюдается повышение мышечного тонуса и рефлексов, вызывающих спастичность. Их руки короткие, с необычными складками на ладонях, с короткими пальцами формы, а аномалии стопы укорочены, пальцы на ногах срослись и обычно незначительны.
У них отсутствует селезенка, а на гениталиях также могут быть неопущенные яички в диапазоне от легкого до тяжелого, что приводит к женскому полу.
Люди с SFMS имеют тяжелую умственную отсталость. Иногда они проявляют беспокойство, проблемы с поведением, судороги и резкую задержку языкового развития. Они эгоцентричны, у них ограничена способность общаться с окружающими. У них также есть психомоторная отсталость что замедляет мысли и уменьшает физические движения. У них корковая атрофия или дегенерация внешнего слоя головного мозга. Корковая атрофия обычно возникает у пожилых людей.[4]
Генетика
SFMS Х-сцепленное заболевание хромосома Xq13. Х-сцепленная карта болезней человека Х хромосома поскольку этот синдром связан с X-хромосомой, женщины, у которых есть две хромосомы, не страдают, но поскольку мужчины имеют только одну X-хромосому, они с большей вероятностью будут затронуты и покажут полные клинические симптомы. Для этого заболевания требуется только одна копия аномального X-сцепленного гена для проявления синдрома. Поскольку у женщин две Х-хромосомы, влияние одной Х-хромосомы рецессивный а вторая хромосома маскирует пораженную хромосому.[5]
Больные отцы никогда не могут передать это заболевание, связанное с Х-хромосомой, своим сыновьям, но больные отцы могут передать ген, связанный с Х-хромосомой, своим дочерям, у которых есть 50% шанс передать этот вызывающий болезнь ген каждому из ее детей. Поскольку женщины, которые наследовать этот ген не проявляет симптомов, они называются перевозчики. У каждого сына-носителя женщины есть 50% шанс проявить симптомы, но ни у одной из дочерей женщины-носителя не будет никаких симптомов.[4]
Было установлено, что некоторые пациенты с SFMS имеют мутация гена в ATRX на Х-хромосоме, также известный как Xq13 на карте. ATRX это генное заболевание, которое связано с другими формами Х-связанной умственной отсталости, такими как Альфа-талассемия / синдром умственной отсталости, Синдром Карпентера, Синдром Юберга-Марсиди, и соастическая параплегия. Возможно, у пациентов с SFMS есть Альфа-талассемия / синдром умственной отсталости без пострадавших гемоглобин H что приводит к Альфаталассемия / синдром умственной отсталости при традиционно признанном заболевании.[5]
Диагностика
Оценка синдрома Смита-Файнмена-Майерса, как и любой другой умственной отсталости, включает подробный семейный анамнез и физический осмотр, который проверяет психику пациента. Пациенту также делают снимки головного мозга и скелета с помощью компьютерной томографии или рентгена. Они также проводят исследование хромосом и некоторые другие генетические биохимические тесты, чтобы помочь выяснить любые другие причины умственной отсталости.
Диагностика SFMS основана на видимых и измеримых симптомах. До 2000 года не было известно о связи SFMS с каким-либо конкретным геном. По состоянию на 2001 год ученые еще не знают, вовлечены ли другие гены в это редкое заболевание. Общий анализ гена ATRX может оказаться полезным в диагностике SFMS.[6]
Лечение
Лечение обычно основано на отображаемых симптомах. Приступы контролируются с помощью противосудорожное средство медикамент. Для решения проблем с поведением врачи прописывают несколько лекарств и программ модификации поведения, в которых участвуют терапевты и другие виды терапии. Даже если умственная отсталость серьезная, она не сокращает продолжительность жизни пациента и не ухудшается с возрастом.
История
15 сентября 1991 года в Сиднее, Австралия, в Детской больнице принца Уэльского сообщалось о двух братьях с отчетливой внешностью, тяжелой умственной отсталостью, низким ростом, крипторхизм (неопущенное яичко), аспления в одном (отсутствие селезенки), резкое нарушение роста, раннее гипотония, а затем и гипертонус, что наводит на мысль о синдроме Смита – Файнмана – Майерса. Все пять зарегистрированных случаев были мужчинами, что позволяет предположить Х-связанный наследование.[7]
23 сентября 1998 года в Исследовательском центре больничных травм и реабилитации Университета Сан-Паулу в Бауру, Бразилия, сообщают о двух мальчиках: монозиготный близнецы, рожденные от нормальных и не кровных родителей, с необычным внешним видом, корковым атрофия, долихоцефалия, невысокий рост, волчья пасть, микрогнатия, выступающие верхние центральные резцы, двусторонняя линия Сиднея, незначительные деформации стопы, неустойчивость при ходьбе, ранняя гипотония, гиперрефлексия, гиперактивность, психомоторная отсталость и серьезная задержка языкового развития. Эти симптомы напоминают симптомы, описанные ранее при синдроме Смита-Файнмана-Майерса.[8]
Рекомендации
- ^ Онлайн-менделевское наследование в человеке (OMIM): 309580
- ^ Согье-Вебер П., Абади В., Монкла А. и др. (Июнь 1993 г.). «Синдром Юберга-Марсиди отображается на проксимальном длинном плече X-хромосомы (Xq12-q21)». Американский журнал генетики человека. 52 (6): 1040–5. ЧВК 1682258. PMID 8503439.
- ^ http://www.whonamedit.com/synd.cfm/2227.html[требуется полная цитата ]
- ^ а б http://health.yahoo.net/galecontent/smith-fineman-myers-syndrome[требуется полная цитата ]
- ^ а б http://www.bookrags.com/research/smith-fineman-myers-syndrome-wog/[требуется полная цитата ]
- ^ http://www.rightdiagnosis.com/s/smith_fineman_myers_syndrome_1/tests.htm[требуется полная цитата ]
- ^ Адес Л.С., Керр Б., Тернер Дж., Мудрый Дж. (Сентябрь 1991 г.). «Синдром Смита-Файнмана-Майерса у двух братьев». Американский журнал медицинской генетики. 40 (4): 467–70. Дои:10.1002 / ajmg.1320400419. PMID 1684092.
- ^ Гион-Алмейда М.Л., Табит А., Кокицу-Наката Н.М., Зечи Р.М. (сентябрь 1998 г.). «Синдром Смита-Файнмана-Майерса у явно монозиготных близнецов». Американский журнал медицинской генетики. 79 (3): 205–8. Дои:10.1002 / (SICI) 1096-8628 (19980923) 79: 3 <205 :: AID-AJMG11> 3.0.CO; 2-L. PMID 9788563.
внешняя ссылка
| Классификация | |
|---|---|
| Внешние ресурсы |