Альфа-талассемия - Alpha-thalassemia
| Альфа-талассемия | |
|---|---|
| Другие имена | α-талассемия |
 | |
| Тип наследования альфа-талассемии | |
| Специальность | Гематология |
| Симптомы | Желтуха, утомляемость[1] |
| Причины | Делеции хромосомы 16р.[2] |
| Диагностический метод | Электрофорез гемоглобина[3] |
| Уход | Переливание крови, возможна спленэктомия[1][4] |
Альфа-талассемия (α-талассемия, α-талассемия) является формой талассемия с участием генов HBA1[5] и HBA2.[6] Талассемии - это группа унаследованный состояние крови что приводит к ухудшению производства гемоглобин, молекула, которая переносит кислород в крови.[7] Нормальный гемоглобин состоит из двух альфа-цепи и два бета-цепочки; при альфа-талассемии наблюдается количественное уменьшение количества альфа-цепей, в результате чего остается меньше нормальных молекул гемоглобина. Кроме того, альфа-талассемия приводит к производству нестабильных молекул бета-глобина, которые вызывают повышенное эритроцит разрушение. Степень нарушения зависит от того, какое клиническое фенотип присутствует (сколько генов поражено).[3]
Признаки и симптомы
Представления людей с альфа-талассемией включают:
| Общий | Необычный |
|---|---|
|
|
Причина
Альфа-талассемии чаще всего передаются по наследству Менделевский рецессивный манера. Они также связаны с удалением хромосома 16p.[2] Альфа-талассемия также может быть приобретена в редких случаях.[12]
Патофизиология
Механизм показывает, что альфа-талассемия приводит к снижению продукции альфа-глобина, следовательно, образуется меньше цепей альфа-глобина, что приводит к избытку β-цепей у взрослых и избытку γ-цепей у новорожденных. Избыточные β-цепи образуют нестабильные тетрамеры, называемые гемоглобин H или HbH четырех бета-цепочки. Избыточные γ-цепи образуют тетрамеры, которые являются плохими переносчиками O2 поскольку их близость к O2 слишком высока, поэтому не диссоциирует на периферии. Гомозигота α0 талассемии, где многочисленные γ4 но никакие α-глобины не встречаются вообще (называемые Hb Barts ), часто приводят к смерти вскоре после рождения.[3][1][13]
Диагностика
Диагноз альфа-талассемии ставится в первую очередь на основании лабораторных исследований и молекулярная диагностика. Альфа-талассемию можно принять за железодефицитная анемия на общий анализ крови или мазок крови, так как оба условия имеют микроцитарную анемию. Железо в сыворотке и сывороточный ферритин может использоваться для исключения железодефицитной анемии.[3]
Типы
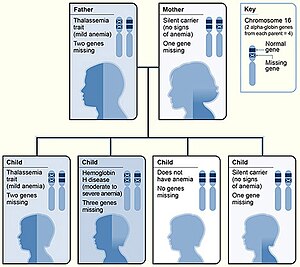
Для α-глобина существует два генетических локуса, таким образом, четыре аллеля находятся в диплоидных клетках. Два аллеля являются материнскими, а два - отцовскими. Тяжесть α-талассемии коррелирует с количеством пораженных аллелей α-глобина: чем больше, тем тяжелее будут проявления болезни. Отмечая генотип, «α» указывает на функциональную альфа-цепь, и '-' патологический.[1][13]
| Затронуты аллели | Описание | Генотип | |||||
|---|---|---|---|---|---|---|---|
| Один | Это известно как альфа-талассемия тихая и с этим типом влияние на синтез гемоглобина минимально. Трех α-глобиновых генов достаточно, чтобы обеспечить нормальное производство гемоглобина и отсутствие клинических симптомов. Это происходит из-за делеционной или неделательной мутации.[1] | - α / α α | |||||
| Два |  Гематопоэз (производство клеток крови) Это состояние называется альфа-талассемией; два альфа-гена допускают почти нормальный производство красных кровяных телец, но мягкий микроцитарный гипохромный анемия виден. Заболевание в такой форме можно принять за железодефицитная анемия и неадекватно обработали железом.[3][1] Признак альфа-талассемии может существовать в двух формах:[1]
| - - / α α или же - α / - α | |||||
| Три | Это состояние называется болезнь гемоглобина H; в крови присутствуют два нестабильных гемоглобина; гемоглобин Бартс (тетрамерный γ цепи ) и гемоглобин H (тетрамерный β цепи ). Оба этих нестабильных гемоглобина имеют более высокое сродство к кислороду, чем нормальный гемоглобин.[14] Микроцитарная гипохромная анемия с клетки-мишени и Тела Heinz (осажденный HbH) на мазок периферической крови может произойти, а также гепатоспленомегалия. Заболевание замечают в детстве или в раннем взрослом возрасте; отмечаются анемия и гепатоспленомегалия.[требуется медицинская цитата ] | - - / - α | |||||
| Четыре | Это известно как большая альфа-талассемия; эти плоды отечный, имеют мало циркулирующего гемоглобина, а гемоглобин, который присутствует, представляет собой все тетрамерные γ-цепи. Когда затронуты все четыре аллеля, плод скорее всего, не переживет беременности без в утробе вмешательство; большинство младенцев с большой альфа-талассемией рождаются мертворожденными с водянка плода. Плоды лечили внутриматочный переливания на протяжении всей беременности, начиная с раннего гестационного возраста, могут дожить до родов с приемлемой заболеваемостью. После рождения варианты лечения включают трансплантацию костного мозга или продолжение хронических переливаний.[15] | - -/- - | |||||
| α α / α α = нормальный: «α α» перед «/» представляет одну хромосому, а «α α» после «/» - ее гомологичная хромосома. | |||||||
Лабораторная диагностика
Первоначальный лабораторный диагноз должен включать: полный анализ крови и показатели эритроцитов.[8] Также мазок периферической крови следует внимательно изучить.[8]
Анализ гемоглобина важен для диагностики альфа-талассемии, поскольку он определяет типы и процентное содержание типов гемоглобина.[16] Существует несколько различных методов анализа гемоглобина, в том числе электрофорез гемоглобина, капиллярный электрофорез и высокоэффективная жидкостная хроматография.[16]
Молекулярный анализ последовательностей ДНК (Анализ ДНК ) может использоваться для подтверждения диагноза альфа-талассемия, особенно для выявления носителей альфа-талассемии (делеции или мутации только в одном или двух альфа-глобин гены).[16]
Уход
Лечение альфа-талассемии может включать: переливание крови для поддержания гемоглобина на уровне, уменьшающем симптомы анемии. Решение о начале переливания зависит от клинической тяжести заболевания.[17] Спленэктомия является возможным вариантом лечения для повышения уровня общего гемоглобина в случаях обострения анемии из-за сверхактивной или увеличенной селезенки или когда трансфузионная терапия невозможна.[18] Однако спленэктомию следует избегать, когда доступны другие варианты, из-за повышенного риска серьезных инфекций и тромбоз.[18]
Кроме того, камни в желчном пузыре может возникнуть проблема, которая потребует хирургического вмешательства. Вторичные осложнения от фебрильный следует контролировать эпизод, и большинство людей живут без лечения.[1][4]
Кроме того, трансплантация стволовых клеток следует рассматривать как лечение (и лекарство), которое лучше всего проводить в раннем возрасте. Другие варианты, например генная терапия, все еще разрабатываются.[19]
Исследование Kreger et al, объединяющее ретроспективный обзор трех случаев большой альфа-талассемии и обзор литературы 17 случаев, показало, что внутриутробное переливание крови может привести к благоприятным результатам. В конечном итоге успешная трансплантация гемопоэтических клеток была проведена у четырех пациентов.[20]
Эпидемиология

Распространение наследственной альфа-талассемии во всем мире соответствует областям малярия воздействие, предполагающее защитную роль. Таким образом, альфа-талассемия распространена в странах к югу от Сахары. Африка, то Средиземноморский бассейн, и вообще тропические (и субтропические) регионы. Эпидемиология альфа-талассемии в США отражает эту глобальную модель распространения. Более конкретно, болезнь HbH проявляется в Юго-Восточная Азия и Средний Восток, в то время как Hb Bart hydrops fetalis признан только в Юго-Восточной Азии.[21]Данные показывают, что 15% Греческий и Турки-киприоты являются носителями бета-талассемия гены, а 10% населения несут гены альфа-талассемии.[22]
Смотрите также
Рекомендации
- ^ а б c d е ж грамм час Орига, Рафаэлла; Мои, Паоло; Галанелло, Ренцо; Цао, Антонио (1 января 1993 г.). «Альфа-талассемия». GeneReviews. PMID 20301608. Получено 22 сентября 2016.обновление 2013
- ^ а б BRS Патология (4-е изд.). Липпинкотт Уильямс и Уилкинс медицинский. Декабрь 2009 г. с. 162. ISBN 978-1451115871.
- ^ а б c d е «Обследование при альфа-талассемии: подходы, лабораторные исследования, электрофорез гемоглобина». emedicine.medscape.com. Получено 24 мая 2016.
- ^ а б «Осложнения и лечение | Талассемия | Заболевания крови | NCBDDD | CDC». www.cdc.gov. Получено 22 сентября 2016.
- ^ Онлайн-менделевское наследование в человеке (OMIM): Гемоглобин - Альфа-локус 1; HBA1 - 141800
- ^ Онлайн-менделевское наследование в человеке (OMIM): Гемоглобин - Альфа-локус 2; HBA2 - 141850
- ^ Руководство Ланжковского по детской гематологии и онкологии, 6-е издание (2016 г.).
- ^ а б c d е «Альфа-талассемия - Симптомы, диагностика и лечение | BMJ Best Practice». bestpractice.bmj.com. Получено 17 ноября 2019.
- ^ Справка, Дом генетики. «Альфа-талассемия». Домашний справочник по генетике. Получено 25 ноября 2019.
- ^ Орига, Рафаэлла; Мои, Паоло (1993), Адам, Маргарет П .; Ardinger, Holly H .; Пагон, Роберта А .; Уоллес, Стефани Э. (ред.), «Альфа-талассемия», GeneReviews®, Вашингтонский университет, Сиэтл, PMID 20301608, получено 25 ноября 2019
- ^ «Оценка анемии - Этиология | Лучшие практики BMJ». bestpractice.bmj.com. Получено 25 ноября 2019.
- ^ Стинсма Д.П., Гиббонс Р.Дж., Хиггс Д.Р. (январь 2005 г.). «Приобретенная альфа-талассемия в сочетании с миелодиспластическим синдромом и другими гематологическими злокачественными новообразованиями». Кровь. 105 (2): 443–52. Дои:10.1182 / кровь-2004-07-2792. PMID 15358626.
- ^ а б Галанелло Р., Цао А. (февраль 2011 г.). «Обзор генного теста. Альфа-талассемия». Генетика в медицине. 13 (2): 83–8. Дои:10.1097 / GIM.0b013e3181fcb468. PMID 21381239.
- ^ «Болезнь гемоглобина H». Orphanet. Получено 22 сентября 2016.
- ^ Вичинский Е.П. (1 января 2009 г.). «Большая альфа-талассемия - новые мутации, внутриутробное ведение и исходы». Гематология. Американское общество гематологии. Образовательная программа. 2009 (1): 35–41. Дои:10.1182 / asheducation-2009.1.35. PMID 20008180.
- ^ а б c Випракасит, VIP; Экваттанакит, Супачай (1 апреля 2018 г.). «Клиническая классификация, скрининг и диагностика талассемии». Гематологические / онкологические клиники Северной Америки. Талассемия. 32 (2): 193–211. Дои:10.1016 / j.hoc.2017.11.006. ISSN 0889-8588. PMID 29458726.
- ^ "Своевременно". www.uptodate.com. Получено 25 ноября 2019.
- ^ а б Тахер, Али; Мусаллам, Халед; Каппеллини, Мария Доменика, ред. (2017). Рекомендации по ведению нетрансфузионно-зависимой талассемии (NTDT) (2-е изд.). Международный фонд талассемии. стр. 24–32. Получено 5 ноября 2019.
- ^ «Талассемия | Врач | Пациент». Пациент. Получено 22 сентября 2016.
- ^ Kreger EM, Singer ST, Witt RG, Sweeters N, Lianoglou B, Lal A и др. (Декабрь 2016 г.). «Благоприятные исходы после внутриутробного переливания у плодов с большой альфа-талассемией: серия случаев и обзор литературы». Пренатальная диагностика. 36 (13): 1242–1249. Дои:10.1002 / pd.4966. PMID 27862048.
- ^ Хартевельд К.Л., Хиггс Д.Р. (май 2010 г.). «Альфа-талассемия». Журнал редких заболеваний Orphanet. 5 (1): 13. Дои:10.1186/1750-1172-5-13. ЧВК 2887799. PMID 20507641.
- ^ Гематология стала проще. АвторДом. 2013-02-06. ISBN 9781477246511.стр. 246
дальнейшее чтение
- Anie KA, Massaglia P (март 2014 г.). «Психологические методы лечения талассемии». Кокрановская база данных систематических обзоров (3): CD002890. Дои:10.1002 / 14651858.cd002890.pub2. ЧВК 7138048. PMID 24604627.
- Галанелло Р., Цао А. (февраль 2011 г.). «Обзор генного теста. Альфа-талассемия». Генетика в медицине. 13 (2): 83–8. Дои:10.1097 / GIM.0b013e3181fcb468. PMID 21381239.
внешняя ссылка
- «Что такое талассемии? - NHLBI, NIH». www.nhlbi.nih.gov. Получено 15 сентября 2016.
| Классификация | |
|---|---|
| Внешние ресурсы |
| Scholia имеет тема профиль для Альфа-талассемия. |