Хронический миеломоноцитарный лейкоз - Chronic myelomonocytic leukemia
| Хронический миеломоноцитарный лейкоз | |
|---|---|
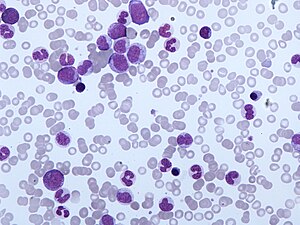 | |
| Мазок периферической крови CMML. Моноцитоз и наличие миелоцитов, метамиелоцитов и промиелоцитов типичны для CMML. | |
| Специальность | Гематология, онкология |
| Причины | Относящийся к окружающей среде канцерогены, ионизирующее излучение, цитотоксические агенты |
| Диагностический метод | Фильм крови, генетическое тестирование |
| Частота | Менее 1 на 100 000 в год |
Хронический миеломоноцитарный лейкоз (CMML) является разновидностью лейкемия, которые раки кроветворных клеток Костный мозг. У взрослых кровяные клетки образуются в костном мозге в результате процесса, известного как кроветворение. В CMML увеличено количество моноциты и незрелые клетки крови (взрывы ) в периферической крови и костном мозге, а также в аномально выглядящих клетках (дисплазия ) по крайней мере в одном типе клеток крови.[1]
CMML показывает характеристики миелодиспластический синдром (МДС); заболевание, при котором клетки крови выглядят ненормально, и миелопролиферативное новообразование (MPN); расстройство, характеризующееся перепроизводством клеток крови. По этой причине в 2002 году CMML был переклассифицирован как расстройство с перекрытием MDS / MPN.[2] Для диагностики CMML Всемирная организация здоровья (ВОЗ) заявляет, что количество моноцитов крови должно быть> 1x10.9/ L, нет Филадельфийская хромосома или мутации в PDGFRA или же PDGFRB ген должен присутствовать, количество бластов должно быть <20% и должна присутствовать дисплазия по крайней мере одной линии миелоидных клеток крови.[3]
Азацитидин это лекарство, используемое для лечения CMML и одобренное Управление по контролю за продуктами и лекарствами (FDA) и Европейское агентство по лекарствам. Пересадка стволовых клеток также используется для лечения CMML и включает трансплантацию донорского гемопоэтические стволовые клетки в получателя. Переливание крови и эритропоэтин используются для лечения заболеваний, связанных анемия.[4][5][6]
Признаки и симптомы
Одним из наиболее распространенных признаков CMML является спленомегалия, встречается примерно в половине случаев. Другие менее частые признаки и симптомы включают: анемия, высокая температура, потеря веса, ночные поты, инфекционное заболевание, кровотечение, синовит, лимфаденопатия, кожная сыпь, плевральный выпот, перикардиальный выпот и перитонеальный выпот.[7][8][9]
Причина
Хотя причина CMML неизвестна, экологические канцерогены, ионизирующее излучение и цитотоксические агенты может играть роль в возникновении болезни.[8] Примерно одна треть случаев МДС с количеством моноцитов> 10% и <1x109/ L перейдет к CMML.[10]
Патогенез
С высокой скоростью Рас мутации в CMML, дерегуляция этого сигнальный путь был связан с патогенезом заболевания. Фактор некроза опухоли, GM-CSF, интерлейкин-3, интерлейкин-4, интерлейкин-6, и интерлейкин-10 может играть роль в гиперпролиферативных клетках CMML. Эти цитокины может стимулировать рост CMML in vitro.[11] Гиперметилирование остатков цитозина (обычно в промоутер регионы гены ) встречается при многих злокачественных новообразованиях, чтобы регулировать экспрессия гена. Одним из обычно гиперметилированных генов CMML является стр. 15INK4b, ген, участвующий в регуляция клеточного цикла.[4]
Генетические мутации
Клональные генетические аномалии распространены при CMML, но они не специфичны для диагностики заболевания. Наиболее частыми являются аномалии 8+, -7 / del (7q) и структурные 12p.[8] KRAS и NRAS мутируют в 25–40% случаев CMML. В Jak2 Мутация V617F обнаруживается в 10% случаев. Мутации в факторах транскрипции, таких как RUNX1, CEBPA, NPM1 и WT1 обнаружены в 30% случаев. Мутации CBL встречаются примерно в 5–18% случаев.[3] Мутации в TET2 гены обнаруживаются примерно в 40–50% CMML.[12] Инактивирующие мутации в одном из двух родительских GATA2 гены приводят к редукции, т.е. гаплонедостаточность, на клеточных уровнях продукта гена GATA2 фактор транскрипции, и тем самым к редкому аутосомно-доминантный генетическое заболевание, GATA2 дефицит. Это заболевание связано с разнообразным набором расстройств, включая миелодиспластический синдром, острый миелоидный лейкоз и CMML. CMML, вызванный дефицитом GATA2, как и другие типы CMML, обычно предшествует моноцитозу.[13][14]
Диагностика
Фильмы крови отображать ряд отклонений. Количество моноцитов> 1x109/ L необходим для диагностики CMML. Другие функции могут включать; лейкоцитоз (50% случаев); сдвиг влево и дисплазия моноцитов и гранулоциты; присутствие метамиелоциты, миелоциты и промоноциты; моноциты с гиперсегментированными ядрами / ядрами аномальной формы, повышенной цитоплазматической базофилией и / или наличием цитоплазматических гранул; эозинофилия (в случаях ХММЛ с эозинофилией); и сфероцитоз (в случаях прямой тест Кумбса, DCT, положительный гемолитическая анемия ). Тромбоцит количество может быть уменьшено, увеличено или нормальным.[7][15][16] Гемоглобин уровни обычно снижаются нормоцитарными и нормохромными эритроцитами. Аутоантитела и холодные агглютинины может присутствовать, и 10% CMML является DCT-положительным.[7][9]Аспираты костного мозга будет демонстрировать гиперцеллюлярность с увеличением количества гранулоцитарных и моноцитарных клеток.[1] Биопсия костного мозга может показать преобладание миелоцитарных и моноцитарных клеток, аномальную локализацию незрелых предшественников и диспластическую мегакариоциты.[1] Моноцитарные узелки - обычное явление при биопсии.[16]
Фенотипические характеристики CMML: CD11b, CD11c, CD14, CD33, CD45 и CD64 наблюдается в 100% случаев; CD13 встречается в 95% случаев; CD4 встречается в 76% случаев; HLA-DR встречается в 71% случаев; CD56 встречается в 53% случаев; CD2 встречается в 34% случаев; CD16 встречается в 29% случаев; CD10 встречается в 28% случаев; CD23 и CD7 встречается в 9% случаев; и CD117 встречается в 5% случаев.[17]
Классификация

Подтипы лейкемии подразделяются на отдельные клинические формы, чтобы их можно было диагностировать и лечить соответствующим образом. Лейкемии подразделяются на лимфоидный и миелоидный новообразования, в зависимости от того, какие клетки костного мозга являются злокачественными. Миелоидные новообразования содержат острые и хронические лейкозы, миелодиспластические синдромы (МДС) и миелопролиферативные новообразования (МПН). MPN характеризуются повышенной продукцией миелоидных клеток крови с более высоким, чем обычно, количеством зрелых клеток. В отличие от MPN, MDS имеют дисфункциональную продукцию миелоидных клеток с пониженным количеством зрелых клеток. Многие из клеток, образующихся при МДС, выглядят ненормально, что известно как дисплазия. CMML показывает характеристики обеих групп, и поэтому заболевание трудно классифицировать.[7][18]
Классификация FAB
В Французско-Американско-Британский (FAB) система классификации лейкозов была опубликована в 1976 году. Он поместил CMML в категорию МДС, наряду с рефрактерной анемией, рефрактерной анемией с кольцевые сидеробласты, рефрактерная анемия с избытком бластов и рефрактерная анемия с избытком бластов в трансформации. Система действительно полезна в клинической практике; однако такие факторы, как цитогенетический статус, не подпадают под классификацию. По этой причине многие болезни в этих группах очень неоднородны.[18][19]
Классификация ВОЗ
В 2001 г. Классификация миелоидных новообразований ВОЗ была опубликована, классифицирующая CMML в новую группу заболеваний, миелодиспластические / миелопролиферативные новообразования (MDS / MPN), что отражает неопластическую природу заболевания. Другие болезни в этой категории: ювенильный миеломоноцитарный лейкоз, атипичный ХМЛ; BCR-ABL1 отрицательный и неклассифицируемый по MDS / MPD. Эти синдромы перекрытия MDS / MPN имеют эффективную продукцию некоторых линий клеток крови, но демонстрируют неэффективную пролиферацию других линий. Пересмотр классификации 2008 г. перенес случаи CMML с транслокациями гена PDGFR в новую группу, миелоидные / лимфоидные новообразования с эозинофилией с аномалиями PDGFRA, PDGFRB или же FGFR1.[2][7][20]
Диагностические критерии
Критерии FAB
Критерии диагностики FAB следующие:[21]
- Количество моноцитов> 1x109/ Л
- 0–19% бластов в костном мозге
- <5% бластов в периферической крови
FAB также произвольно классифицирует CMML на группы миелодиспластического и миелопролиферативного типов. Показатель лейкоцитов 13х109 используется в качестве отсечки, чтобы различать эти два.[12]
Критерии ВОЗ
Критерии постановки диагноза ВОЗ следующие:[3]
- Стойкий моноцитоз периферической крови с числом> 1x109/ Л
- Нет филадельфийской хромосомы или гена слияния BCR-ABL1
- Нет реаранжировки гена PDGFRA или PDGFRB
- <20% миелобластов, монобластов и промоноцитов в периферической крови или костном мозге
- Дисплазия одной или нескольких миелоидных линий; если миелодисплазия отсутствует или минимальна, диагноз ХММЛ может быть поставлен при соблюдении других требований и:
- В гематопоэтических клетках присутствует молекулярно-генетическая аномалия, или
- Моноцитоз в течение ≥3 месяцев и другие причины моноцитоза исключены.
Согласно определению ВОЗ CMML состоит из двух основных подмножеств: CMML-1 и CMML-2. CMML-1 диагностируется, если миелобласты, монобласты и промоноциты составляют <5% периферической крови и <10% костного мозга. CMML-2 диагностируется, если:
- Миелобласты, монобласты или промоноциты составляют 5-19% в крови или
- Миелобласты, монобласты или промоноциты составляют 10-19% в костном мозге, или
- Стержни Ауэра присутствуют
CMML-1 и CMML-2 могут быть дополнительно сгруппированы как CMML-1 или CMML-2 с эозинофилией. Они диагностируются, если соблюдены вышеуказанные критерии и количество эозинофилов в крови> 1,5х10.9/ L.[8]
Наличие двух или более фенотипических аномалий может помочь в диагностике CMML при отсутствии идентификации цитогенетических или диспластических признаков. Они могут включать экспрессию CD56 и / или CD2 или недостаточную экспрессию HLA-DR.[3]
Прогноз
Факторы, влияющие на прогноз
CMML-2 имеет пониженную общую выживаемость по сравнению с CMML-1, при этом медиана выживаемости составляет 15 и 20 месяцев соответственно. Миелопролиферативный CMML (> 13x109 моноцитов / л) имеет меньшую выживаемость по сравнению с миелодиспластическим CMML. Количество тромбоцитов <100 x109/ Л снижает общую выживаемость. Уровень гемоглобина <10 г / дл снижает общую выживаемость. Некоторые цитогенетические нарушения влияют на прогноз CMML. Нормальный кариотип или единичная потеря Y-хромосомы имеют низкий прогноз риска. Трисомия 8, аномалии хромосомы 7 и сложные кариотипы составляют группу высокого риска. Другие цитогенетические аномалии имеют промежуточный прогноз. Соматические мутации в таких генах, как ASXL1 и EZH2 связаны с плохим прогнозом.[12]
CMML имеет 20–30% шанс трансформации в AML, что ниже, чем у других подобных заболеваний. Подтип CMML-2 связан с повышенным риском трансформации, а мутации ASXL1 и RUNX1 также увеличивают риск перехода к AML.[12][22][23]
Системы подсчета очков
IPSS
В Международная прогностическая система оценки (IPSS) был разработан в середине 1990-х годов для оценки прогноза пациентов с МДС. В этой системе случаи делятся на 2 группы; группа низкого риска (подразделяется на низкий и средний-1) и более высокий риск (подразделяется на промежуточный-2 и высокий). Он использует процент бластов, количество цитопений и данные цитогенетики костного мозга, чтобы отнести случаи CMML к этим группам. Из-за системы оценки, разрабатываемой для МДС, большее количество миелопролиферативных случаев CMML (лейкоциты> 13x109) исключаются из системы подсчета очков. Хотя балльная система IPSS используется в клинической практике, в каждой группе наблюдается высокая вариабельность. По этой причине разрабатываются новые методы оценки прогноза при МДС (и ХММЛ).[12][24]
Прогностическая скоринговая система MD Anderson
Новый метод, разработанный с использованием данных из Онкологический центр доктора медицины Андерсона обнаружили, что уровень гемоглобина <12 г / дл, общее количество циркулирующих лимфоцитов> 2,5 x 109/ Л,> 0% незрелых миелоидных клеток,> 10% бластов костного мозга вызывает снижение общей выживаемости. Эти данные позволяют разделить случаи CMML на группы низкого, среднего 1, среднего 2 и высокого риска. Эти группы имеют среднее время выживания 24, 15, 8 и 5 месяцев соответственно.[25][26]
Дюссельдорфская партитура
Шкала Дюссельдорфа разделяет случаи по четырем категориям, давая по одному баллу за каждую; бласты костного мозга ≥5%, ЛДГ> 200 Ед / л, гемоглобин ≤9 г / дл и количество тромбоцитов ≤100000 / мкл. Оценка 0 указывает на группу низкого риска, 1-2 указывает на группу среднего риска, а 3-4 указывает на группу высокого риска. Кумулятивная 2-летняя выживаемость с оценками 0, 1-2 и 3-4 составляет 91%, 52% и 9%; и риск трансформации AML составляет 0%, 19% и 54% соответственно.[10]
Уход
Лечение CMML остается сложной задачей из-за отсутствия клинических испытаний, изучающих болезнь как отдельную клиническую сущность. В клинических испытаниях его часто объединяют с МДС, и по этой причине лечение ХММЛ очень похоже на лечение МДС. Большинство случаев рассматриваются как поддерживающие, а не лечебные, потому что большинство методов лечения не обеспечивают эффективного увеличения выживаемости. Показания к лечению включают наличие B симптомы, симптоматическое поражение органов, повышение показателей крови, гиперлейкоцитоз, лейкостаз и / или ухудшение цитопении.[6][10]
Переливание крови и эритропоэтин Применяются для повышения уровня гемоглобина в случаях анемии.[6]
Азацитидин это лекарство, одобренное США. Управление по контролю за продуктами и лекарствами (FDA) для лечения CMML и Европейское агентство по лекарствам (EMA) для нераспространения CMML высокого риска с 10–19% бластов костного мозга. Это аналог цитидина, который вызывает гипометилирование ДНК путем ингибирования ДНК-метилтрансфераза. Децитабин является препаратом, аналогичным азацитидину, и одобрен FDA для лечения всех подтипов МДС, включая CMML. Гидроксимочевина это химиотерапия, которая используется при миелопролиферативной форме CMML для уменьшения количества клеток.[4][10][12] Децитабин / кедазуридин (Inqovi) - это фиксированная комбинированный препарат для лечения взрослых с миелодиспластическими синдромами (МДС) и хроническим миеломоноцитарным лейкозом (ХММЛ), который был одобрен для использования в США в июле 2020 года.[27]
Трансплантация гемопоэтических стволовых клеток остается единственным лечебным средством от CMML. Однако из-за позднего возраста начала и наличия других заболеваний такая форма лечения часто невозможна.[5][28]
Эпидемиология
Было несколько отдельных эпидемиологический исследования CMML из-за сложности классификации заболевания. Заболеваемость CMML составляет менее 1 на 100 000 человек в год.[12]Средний возраст постановки диагноза 65–75 лет. CMML больше подходит мужчинам, чем женщинам, в соотношении 1,5–3: 1.[8]
Рекомендации
- ^ а б c Фукар К. (август 2009 г.). «Миелодиспластические / миелопролиферативные новообразования». Являюсь. J. Clin. Патол. 132 (2): 281–9. Дои:10.1309 / AJCPJ71PTVIKGEVT. PMID 19605822.
- ^ а б Вардиман Дж. У., Харрис Н. Л., Брунинг Р. Д. (октябрь 2002 г.). «Классификация миелоидных новообразований Всемирной организации здравоохранения (ВОЗ)». Кровь. 100 (7): 2292–302. Дои:10.1182 / кровь-2002-04-1199. PMID 12239137.
- ^ а б c d Вардиман Дж, Хиек Э (2011). «Классификация, оценка и генетика вариантов миелопролиферативного новообразования Всемирной организации здравоохранения». Гематология. 2011: 250–6. Дои:10.1182 / asheducation-2011.1.250. PMID 22160042.
- ^ а б c McCormack, SE; Уорлик, ED (7 сентября 2010 г.). «Эпигенетические подходы в лечении миелодиспластических синдромов: клиническое применение азацитидина». ОнкоЦели и терапия. 3: 157–65. Дои:10.2147 / OTT.S5852. ЧВК 2939768. PMID 20856790.
- ^ а б Роберт Дж. Сойфер (17 ноября 2008 г.). Трансплантация гемопоэтических стволовых клеток. Springer. ISBN 978-1-934115-05-3. Получено 23 сентября 2012.
- ^ а б c Беннетт, Дж. М. (июнь 2002 г.). «Хронический миеломоноцитарный лейкоз». Современные варианты лечения в онкологии. 3 (3): 221–3. Дои:10.1007 / s11864-002-0011-6. PMID 12057067.
- ^ а б c d е Бэйн, Барбара Дж. (2003). Диагностика лейкемии. Кембридж, Массачусетс: издательство Blackwell Publishers. ISBN 978-1-4051-0661-0.
- ^ а б c d е Патология и генетика гемо (Классификация опухолей Всемирной организации здравоохранения S.). Oxford Univ Pr. 2003 г. ISBN 978-92-832-2411-2.
- ^ а б Пол Мосс; Виктор Хоффбранд (2011). Essential Hematology, включает БЕСПЛАТНУЮ версию для ПК (Essentials). Вили-Блэквелл. ISBN 978-1-4051-9890-5.
- ^ а б c d Виктория Фабер; Ричард Грейл; Лиза Плейер; Даниэль Нойрайтер (2010). Хронические миелоидные неоплазии и синдромы клонального перекрытия: эпидемиология, патофизиология и варианты лечения. Берлин: Springer. ISBN 978-3-211-79891-1.
- ^ Арчечи Р.Дж., Лонгли Б.Дж., Эмануэль П.Д. (2002). «Атипичные клеточные нарушения». Гематология. 2002: 297–314. Дои:10.1182 / asheducation-2002.1.297. PMID 12446429.
- ^ а б c d е ж грамм Каццола М., Мальковати Л., Инверницци Р. (2011). «Миелодиспластические / миелопролиферативные новообразования». Гематология. 2011: 264–72. Дои:10.1182 / asheducation-2011.1.264. PMID 22160044. S2CID 24489846.
- ^ Криспино Дж. Д., Хорвиц М. С. (апрель 2017 г.). «Мутации фактора GATA при гематологических заболеваниях». Кровь. 129 (15): 2103–2110. Дои:10.1182 / кровь-2016-09-687889. ЧВК 5391620. PMID 28179280.
- ^ Хирабаяши С., Влодарски М.В., Козыра Е., Нимейер С.М. (август 2017 г.). «Гетерогенность миелоидных новообразований, связанных с GATA2». Международный журнал гематологии. 106 (2): 175–182. Дои:10.1007 / s12185-017-2285-2. PMID 28643018.
- ^ Hirschmann, Jan V .; Ткачук, Дуглас С .; Винтроб, Максвелл Майер (2007). Атлас клинической гематологии Винтроба. Филадельфия: Wolters Kluwer Health / Lippincott Williams & Wilkins. ISBN 978-0-7817-7023-1.
- ^ а б Уэйн В. Гроди; Наим, Фарамарц (2008). Гематопатология: морфология, иммунофенотип, цитогенетика и молекулярные подходы. Амстердам: Elsevier / Academic Press. ISBN 978-0-12-370607-2.
- ^ Foxwell Натан Эммонс; Войцех Горчица; Джеймс Вайсбергер (2004). Атлас дифференциальной диагностики в неопластической гематопатологии. Вашингтон, округ Колумбия: Тейлор и Фрэнсис. ISBN 978-1-84214-247-9.
- ^ а б Бхаргава Р., Далал Б.И. (2010). «Два шага вперед, один шаг назад: 4-я классификация миелоидных новообразований ВОЗ (2008 г.)». Индийский J Pathol Microbiol. 53 (3): 391–4. Дои:10.4103/0377-4929.68240. PMID 20699489.
- ^ Turgeon, Мэри Луиза (1999). Клиническая гематология: теория и процедуры. Хагерствон, доктор медицины: Липпинкотт Уильямс и Уилкинс. С. 321–322. ISBN 978-0-316-85623-2.
- ^ Цонгалис, Грегори Дж .; Коулман, Уильям Л. (2009). МОЛЕКУЛЯРНАЯ ПАТОЛОГИЯ: МОЛЕКУЛЯРНАЯ ОСНОВА ЗАБОЛЕВАНИЙ ЧЕЛОВЕКА; ED. УИЛЬЯМ Б. КОЛЕМАН. Амстердам: Elsevier Academic Press. ISBN 978-0-12-374419-7.
- ^ Беннетт Дж. М., Катовски Д., Дэниел М. Т. и др. (Июнь 1982 г.). «Предложения по классификации миелодиспластических синдромов». Br. J. Haematol. 51 (2): 189–99. CiteSeerX 10.1.1.630.8355. Дои:10.1111 / j.1365-2141.1982.tb02771.x. PMID 6952920.
- ^ Уильям Г. Финн; ЛоЭнн С. Петерсон (31 мая 2004 г.). Гематопатология в онкологии. Springer. С. 33–. ISBN 978-1-4020-7919-1. Получено 23 сентября 2012.
- ^ Барбара Дж. Бэйн (2003). Хронические миелопролиферативные заболевания: цитогенетические и молекулярно-генетические аномалии. Karger Publishers. С. 72–. ISBN 978-3-8055-7307-8. Получено 23 сентября 2012.
- ^ Гринберг П., Кокс С., ЛеБо М.М. и др. (Март 1997 г.). «Международная балльная система оценки прогноза миелодиспластических синдромов». Кровь. 89 (6): 2079–88. Дои:10.1182 / blood.V89.6.2079. PMID 9058730.
- ^ Гарсия-Манеро Джи (2010). «Прогноз миелодиспластических синдромов». Гематология. 2010: 330–7. Дои:10.1182 / asheducation-2010.1.330. PMID 21239815.
- ^ Онида Ф., Кантарджиан Х.М., Смит Т.Л. и др. (Февраль 2002 г.). «Прогностические факторы и системы оценки при хроническом миеломоноцитарном лейкозе: ретроспективный анализ 213 пациентов». Кровь. 99 (3): 840–9. Дои:10.1182 / кровь.V99.3.840. PMID 11806985. S2CID 1310629.
- ^ «FDA одобрило новую терапию миелодиспластических синдромов (МДС), которую можно проводить дома». НАС. Управление по контролю за продуктами и лекарствами (FDA) (Пресс-релиз). 7 июля 2020. Получено 7 июля 2020.
 Эта статья включает текст из этого источника, который находится в всеобщее достояние.
Эта статья включает текст из этого источника, который находится в всеобщее достояние. - ^ Bacher U, Haferlach T, Schnittger S, Kreipe H, Kröger N (март 2011 г.). «Последние достижения в диагностике, молекулярной патологии и терапии хронического миеломоноцитарного лейкоза». Br J Haematol. 153 (2): 149–67. Дои:10.1111 / j.1365-2141.2011.08631.x. PMID 21401573.
внешняя ссылка
| Классификация | |
|---|---|
| Внешние ресурсы |