Филадельфийская хромосома - Philadelphia chromosome
| Филадельфийская хромосома | |
|---|---|
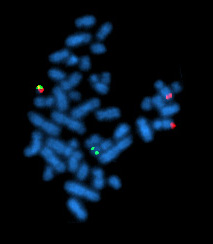 | |
| Метафазная клетка, положительная по перегруппировке bcr / abl с использованием РЫБЫ | |
| Специальность | Онкология |
В Филадельфийская хромосома или же Филадельфия транслокация (Ph) является специфической генетической аномалией в хромосома 22 из лейкозный рак клетки (особенно хронический миелоидный лейкоз (CML) клетки). Эта хромосома дефектная и необычно короткая из-за реципрокная транслокация, t (9; 22) (q34; q11), генетического материала между хромосома 9 и хромосома 22, и содержит ген слияния называется BCR-ABL1. Этот ген является ABL1 ген хромосомы 9 наложен на область кластера точки останова BCR ген хромосомы 22, кодирование для гибридного белка: a тирозинкиназа сигнализация белок, который "всегда включен", заставляя клетку разделять неконтролируемо, нарушая стабильность генома и нарушая различные сигнальные пути, управляющие клеточным циклом.[1]
Наличие этой транслокации необходимо для диагностики ХМЛ; другими словами, все случаи CML положительны для BCR-ABL1.[2] (Некоторые случаи затруднены загадочной транслокацией, невидимой на G-полосный препараты хромосом или вариант транслокации с участием другой хромосомы или хромосом, а также длинного плеча хромосом 9 и 22. Другие похожие, но действительно Ph-отрицательные состояния считаются CML-подобными миелопролиферативными новообразованиями.[3]) Однако наличие филадельфийской (Ph) хромосомы недостаточно. специфический для диагностики ХМЛ, так как он также встречается в острый лимфобластный лейкоз[4] (также известный как ОЛЛ, 25–30% случаев у взрослых и 2–10% случаев педиатрический случаях) и иногда в острый миелолейкоз (AML), а также острый лейкоз смешанного фенотипа (MPAL).
Молекулярная биология

Хромосомный дефект в филадельфийской хромосоме является взаимным перемещение, в котором части двух хромосом, 9 и 22, меняются местами. В результате ген слияния создается путем сопоставления ABL1 ген на хромосоме 9 (область q34) в часть BCR (область кластера точки останова) ген на хромосома 22 (регион q11). Это реципрокная транслокация, создающая удлиненную хромосому 9 (называемую производной хромосомой или der 9), и усеченная хромосома 22 (Филадельфийская хромосома, 22q-).[5][6] В соответствии с Международная система цитогенетической номенклатуры человека (ISCN), это хромосомная транслокация обозначается как t (9; 22) (q34; q11). Символ ABL1 происходит от Абельсон, имя вирус лейкемии который несет аналогичный белок. Символ BCR происходит от области кластера точки разрыва, гена, который кодирует белок, который действует как фактор обмена гуаниновых нуклеотидов для белков Rho GTPase. [7]
Транслокация приводит к онкогенный Слитый ген BCR-ABL1, который можно найти на более короткой производной хромосоме 22. Этот ген кодирует слитый белок BCR-ABL1. В зависимости от точного места слияния молекулярная масса этого белка может составлять от 185 до 210. кДа. Следовательно, гибридный слитый белок BCR-ABL1 обозначается как p210 или p185.
Три клинически важных варианта, кодируемых гибридным геном, представляют собой изоформы p190, p210 и p230.[8] p190 обычно связан с B-клетками острый лимфобластный лейкоз (ВСЕ), а p210 обычно ассоциируется с хронический миелоидный лейкоз но также может быть связан с ВСЕ и ПОД.[9] p230 обычно связан с хроническим миелолейкозом, связанным с нейтрофилией и тромбоцитозом (CML-N).[9] Кроме того, изоформа p190 также может быть выражена как вариант сращивания из p210.[10]
Ген ABL1 экспрессирует ассоциированный с мембраной белок, a тирозинкиназа, а транскрипт BCR-ABL1 также транслируется в тирозинкиназу, содержащую домены как из генов BCR, так и из ABL1. Активность тирозинкиназ обычно регулируется аутоингибиторным образом, но слитый ген BCR-ABL1 кодирует белок, который «всегда включен» или конститутивно активирован, что приводит к нарушению связывания ДНК и нерегулируемому делению клеток (то есть раку). Это происходит из-за замены миристоилированной кэп-области, которая, когда она присутствует, вызывает конформационное изменение, делающее киназный домен неактивным, на усеченную часть белка BCR.[11] Хотя область BCR также экспрессирует серин / треонинкиназы, тирозинкиназа функция очень актуальна для медикаментозной терапии. Поскольку N-концевые домены Y177 и CC из BCR кодируют конститутивную активацию киназы ABL1, эти области нацелены в терапии для подавления активности киназы BCR-ABL1. Ингибиторы тирозинкиназы специфичны для таких доменов, как CC, Y177 и Rho (например, иматиниб и сунитиниб ) являются важными препаратами против различных видов рака, включая ХМЛ, карцинома почек (RCC) и опухоли стромы желудочно-кишечного тракта (ГИСО).
Слитый BCR-ABL1 белок взаимодействует с рецептор интерлейкина-3 бета (c) субъединица и модерируется цикл активации внутри его домена SH1, который включается при связывании с АТФ и запускает нисходящие пути. Активность тирозинкиназы ABL1 BCR-ABL1 повышен по сравнению с ABL1 дикого типа.[12] Поскольку ABL активирует ряд клеточный цикл -контроль белки и ферменты, результат BCR-ABL1 слияние должно ускорить деление клеток. Более того, он подавляет Ремонт ДНК, вызывая геномная нестабильность и потенциально вызывая опасения взрывной кризис в CML.
Пролиферативная роль при лейкемии
Ген слияния BCR-ABL1 и белок, кодируемый филадельфийской хромосомой, воздействуют на несколько сигнальных путей, которые напрямую влияют на апоптотический потенциал, скорость деления клеток и различные стадии клеточного цикла для достижения неконтролируемой пролиферации, характерной для CML и ALL.
Путь JAK / STAT
Особенно важна передача сигналов цитокинов и факторов роста для выживания и пролиферации клеток миелогенного лейкоза в микроокружении костного мозга. В JAK / STAT Путь регулирует многие из этих эффекторов путем активации STAT, которые являются факторами транскрипции со способностью модулировать рецепторы цитокинов и факторы роста. JAK2 фосфорилирует слитый белок BCR-ABL по Y177 и стабилизирует слитый белок, усиливая передачу сигналов онкогенных клеток. Было показано, что мутации JAK2 являются центральными для миелопролиферативных новообразований, а киназы JAK играют центральную роль в развитии гематологических злокачественных новообразований (журнал крови JAK). Терапия ALL и CML нацелена на JAK2, а также на BCR-ABL с использованием нилотиниб и руксолитиниб в мышиных моделях для подавления нижестоящей передачи сигналов цитокинов путем подавления активации транскрипции STAT3 и STAT5 (appelmann et al). Взаимодействие между JAK2 и BCR-ABL внутри этих гематопоэтических злокачественных новообразований подразумевает важную роль JAK-STAT-опосредованной передачи сигналов цитокинов в стимулировании роста лейкозных клеток, проявляющих активность Ph-хромосомы и тирозинкиназы BCR-ABL. Хотя центральная роль пути JAK2 в прямой пролиферации при CML обсуждается, его роль в качестве нижестоящего эффектора тирозинкиназы BCR-ABL сохраняется. Воздействие на клеточный цикл через JAK-STAT в основном периферическое, но, непосредственно влияя на поддержание гематопоэтической ниши и окружающей ее микросреды, активация BCR-ABL передачи сигналов JAK-STAT играет важную роль в поддержании роста и деления лейкемических клеток.[13][14]
Путь Ras / MAPK / ERK
В Рас / МАПК / ЭРК Путь передает сигналы ядерным факторам транскрипции и играет роль в управлении контролем клеточного цикла и дифференцировке. В клетках, содержащих Ph-хромосому, тирозинкиназа BCR-ABL активирует путь RAS / RAF / MEK / ERK, что приводит к нерегулируемой пролиферации клеток через транскрипцию генов в ядре. Тирозинкиназа BCR-ABL активирует Ras посредством фосфорилирования белка GAB2, которое зависит от BCR-локализованного фосфорилирования Y177. В частности, показано, что Ras является важной нижестоящей мишенью для BCR-ABL1 при ХМЛ, поскольку мутанты Ras на мышиных моделях нарушают развитие CML, связанного с геном BCR-ABL1 (эффект ингибирования Ras в гемопоэзе и лейкемогенезе BCR / ABL). Путь Ras / RAF / MEK / ERK также участвует в сверхэкспрессии остеопонтин (OPN), который важен для поддержания ниши гемопоэтических стволовых клеток, что косвенно влияет на неконтролируемую пролиферацию, характерную для лейкозных клеток. Клетки слияния BCR-ABL также демонстрируют постоянно высокие уровни активированного Ras, связанного с GTP, активируя Ras-зависимый сигнальный путь, который, как было показано, ингибирует апоптоз ниже BCR-ABL (Cortez et al). Взаимодействие с рецептором IL-3 также индуцирует путь Ras / RAF / MEK / ERK для фосфорилирования факторов транскрипции, которые играют роль в управлении переходом G1 / S клеточного цикла.[15][16][17]
Связывание ДНК и апоптоз
Ген c-Abl в клетках дикого типа участвует в связывании ДНК, что влияет на такие процессы, как транскрипция ДНК, репарация, апоптоз и другие процессы, лежащие в основе клеточного цикла. Хотя природа этого взаимодействия обсуждалась, существуют доказательства того, что c-Abl фосфорилирует HIPK2, серин / треонинкиназа, в ответ на повреждение ДНК и способствует апоптозу в нормальных клетках. Напротив, слияние BCR-ABL ингибирует апоптоз, но его влияние на связывание ДНК, в частности, неясно.[18] Было показано, что при ингибировании апоптоза клетки BCR-ABL устойчивы к лекарственному апоптозу, но также имеют проапоптотический профиль экспрессии за счет повышенных уровней экспрессии p53, p21 и Bax. Однако функция этих проапоптотических белков нарушена, и в этих клетках не осуществляется апоптоз. BCR-ABL также участвует в предотвращении процессинга каспазы 9 и каспазы 3, что усиливает ингибирующий эффект.[19][20][21] Другим фактором, препятствующим прогрессированию клеточного цикла и апоптозу, является делеция гена IKAROS, которая присутствует в> 80% случаев ОЛЛ с положительной Ph-хромосомой. Ген IKAROS имеет решающее значение для остановки клеточного цикла, опосредованной рецептором Pre-B, в ВСЕХ клетках, положительных по Ph, который при нарушении обеспечивает механизм неконтролируемого прогрессирования клеточного цикла и пролиферации дефектных клеток, что стимулируется передачей сигналов тирозинкиназы BCR-ABL.[22]
Номенклатура
Обозначена филадельфийская хромосома Ph (или Ph ') хромосома и обозначает укороченную хромосому 22, которая кодирует гибридный ген BCR-ABL / протеинкиназу. Он возникает из-за транслокации, которую называют t (9; 22) (q34.1; q11.2), между хромосомой 9 и хромосомой 22, с разрывами, происходящими в области (3), полосе (4), поддиапазоне (1) длинного плеча (q) хромосомы 9 и области (1), полосе (1), поддиапазоне -полоса (2) длинного плеча (q) хромосомы 22. Следовательно, точки разрыва хромосомы записываются как (9q34.1) и (22q11.2), соответственно, с использованием стандартов ISCN.
Терапия
Ингибиторы тирозинкиназы

В конце 1990-х СТИ-571 (иматиниб, Gleevec / Glivec) был идентифицирован фармацевтической компанией Новартис (затем известная как Ciba Geigy) в грохотах с высокой пропускной способностью для ингибиторы тирозинкиназы. Последующие клинические испытания под руководством доктора Х. Брайан Дж. Друкер в Орегонский университет здоровья и науки в сотрудничестве с доктором Чарльзом Сойерсом и доктором Моше Талпазом продемонстрировали, что STI-571 ингибирует пролиферацию гематопоэтических клеток, экспрессирующих BCR-ABL. Хотя он не уничтожил клетки ХМЛ, он значительно ограничил рост опухолевого клона и снизил риск появления опасностей "взрывной кризис ".[нужна цитата ] В 2000 г. Джон Куриян определили механизм, с помощью которого STI-571 ингибирует киназный домен Abl.[23] Он был продан в 2001 году компанией Novartis как мезилат иматиниба (Гливек в США, Гливек в Европе).
В настоящее время разрабатываются другие фармакологические ингибиторы, которые являются более сильными и / или активными против появляющихся устойчивых к Gleevec / Glivec клонов BCR-abl у леченных пациентов. Большинство этих устойчивых клонов представляют собой точечные мутации в киназе BCR-abl. Новые ингибиторы включают дазатиниб и нилотиниб, которые значительно более эффективны, чем иматиниб, и могут преодолеть резистентность. Комбинированная терапия с нилотинибом и руксолитнибом также продемонстрировала успех в подавлении резистентности за счет одновременного воздействия на стадии JAK-STAT и BCR-ABL. Ингибиторы малых молекул, например триоксид мышьяка и гельданамицин аналоги, также были идентифицированы в подавлении трансляции киназы BCR-ABL и способствовании ее деградации протеазой.[24][25]
Акситиниб, препарат, используемый для лечения почечно-клеточного рака, показал свою эффективность в подавлении активности киназы Abl у пациентов с BCR-ABL1 (T315I).[26] Мутация T315I в гене слияния придает устойчивость к другим ингибиторам тирозинкиназы, таким как иматиниб, однако акситиниб успешно применялся для лечения пациента с ВСЕ несущие эту мутацию, а также CML клетки в культуре.
Лечение педиатрических Ph + ALL комбинацией стандартных химиотерапия и RTK ингибиторы могут привести к ремиссии,[нужна цитата ] но лечебный потенциал неизвестен.
Пересадка крови или костного мозга
Потенциально лечебным, но рискованным вариантом лечения Ph + ALL или Ph + CML у детей является трансплантация костного мозга или пуповинная кровь трансплантат, но некоторые предпочитают химиотерапию для достижения первой ремиссии (CR1). Для некоторых трансплантат костного мозга от подобранного родственного донора или подобранного неродственного донора может быть предпочтительным при достижении ремиссии.
Пуповинная кровь Некоторые предпочитают трансплантацию, когда соответствие костного мозга 10/10 недоступно, и трансплантация пуповинной крови может иметь некоторые преимущества, в том числе снижение частоты болезни трансплантат против хозяина (РТПХ), которая является частым и значительным осложнением трансплантации. . Однако трансплантат с пуповинной кровью иногда требует более длительных периодов времени для приживления, что может увеличить вероятность осложнений из-за инфекции. Независимо от типа трансплантата возможны летальность и рецидив, связанный с трансплантацией, и показатели могут меняться по мере улучшения протоколов лечения. Для второй ремиссии (CR2), если она достигнута, возможны варианты как химиотерапии, так и трансплантации, и многие врачи предпочитают трансплантацию.[нужна цитата ]
История
Филадельфийская хромосома была впервые обнаружена и описана в 1959 году. Дэвид Хангерфорд из онкологического центра Фокса Чейза (затем - Института онкологических исследований) и Питер Новелл от Медицинский факультет Пенсильванского университета, и назван в честь города, в котором расположены оба объекта.[1][27][28]
Хангерфорд писал докторскую диссертацию по хромосомам в лаборатории генетики онкологического центра Фокса Чейза и обнаружил крошечный дефект в хромосомах в клетках крови пациентов с лейкемией. Это был первый генетический дефект, связанный с определенным раком человека. Ноуэлл был патологом в Пенсильванском университете, изучая лейкозные клетки под микроскопом, когда он заметил клетки в процессе деления. К его удивлению, их хромосомы - обычно нечеткий клубок - были видны как отдельные структуры.
Ноуэлл искал в этом районе специалиста по хромосомам, чтобы работать с ним, и нашел Хангерфорда. Проводя свои микроскопические исследования, Хангерфорд заметил, что некоторые лейкозные клетки имеют аномально короткую хромосому 22. Мутация стала известна как филадельфийская хромосома.
В 1973 г. Джанет Роули на Чикагский университет идентифицировал механизм возникновения филадельфийской хромосомы как транслокации.[1][29][30]
Смотрите также
Рекомендации
- ^ а б c Вапнер Дж. Хромосома Филадельфии: генетическая тайна, смертельный рак и невероятное изобретение спасающего жизнь лечения. ISBN 9781615191970
- ^ "https://www.nccn.org/professionals/physician_gls/pdf/cml.pdf" (PDF). nccn.org. Получено 2020-07-15. Внешняя ссылка в
| название =(помощь) - ^ "https://www.nccn.org/professionals/physician_gls/pdf/mpn.pdf" (PDF). nccn.org. Получено 2018-02-20. Внешняя ссылка в
| название =(помощь) - ^ Талпаз М., Шах Н.П., Кантарджиан Х. и др. (Июнь 2006 г.). «Дазатиниб при устойчивых к иматинибу филадельфийских хромосомных лейкозах». N. Engl. J. Med. 354 (24): 2531–41. Дои:10.1056 / NEJMoa055229. PMID 16775234.
- ^ Kurzrock, R .; Kantarjian, H.M .; Druker, B.J .; Талпаз, М. (2003). «Филадельфийские хромосомно-положительные лейкемии: от основных механизмов к молекулярной терапии». Анналы внутренней медицины. 138 (10): 819–830. Дои:10.7326/0003-4819-138-10-200305200-00010. PMID 12755554.
- ^ Мело, Дж. В. (1996). «Молекулярная биология хронического миелоидного лейкоза». Лейкемия. 10 (5): 751–756. PMID 8656667.
- ^ «Запись гена для BCR». NCBI Gene. Получено 21 января 2020.
- ^ Advani, A. S .; Пендергаст, А. М. (2002). «Варианты Bcr-Abl: биологические и клинические аспекты». Исследование лейкемии. 26 (8): 713–720. Дои:10.1016 / s0145-2126 (01) 00197-7. PMID 12191565.
- ^ а б Pakakasama, S .; Kajanachumpol, S .; Kanjanapongkul, S .; Sirachainan, N .; Meekaewkunchorn, A .; Ningsanond, V .; Хунген, С. (2008). «Простая мультиплексная ОТ-ПЦР для определения общих транскриптов слияния при остром лейкозе у детей». Международный журнал лабораторной гематологии. 30 (4): 286–291. Дои:10.1111 / j.1751-553X.2007.00954.x. PMID 18665825.
- ^ Lichty, B.D .; Китинг, А .; Callum, J .; Yee, K .; Croxford, R .; Corpus, G .; Nwachukwu, B .; Kim, P .; Guo, J .; Камель-Рид С. (1998). «Экспрессия p210 и p190 BCR-ABL из-за альтернативного сплайсинга при хроническом миелолейкозе». Британский журнал гематологии. 103 (3): 711–715. Дои:10.1046 / j.1365-2141.1998.01033.x. PMID 9858221.
- ^ Нагар, Бхушан (21 марта 2003 г.). «Структурная основа аутоингибирования c-Abl тирозинкиназы». Клетка. 112 (6): 859–871. Дои:10.1016 / s0092-8674 (03) 00194-6. PMID 12654251.
- ^ Саттлер, Мартин; Джеймс Д. Гриффин (апрель 2001 г.). «Механизмы трансформации онкогеном BCR / ABL». Международный журнал гематологии. 73 (3): 278–91. Дои:10.1007 / BF02981952. PMID 11345193.
- ^ Варшава, Вт; Вальц, C; Sexl, V (2014). «JAK на все руки: JAK2-STAT5 как новые терапевтические мишени при хроническом миелоидном лейкозе BCR-ABL1». Кровь. 123 (19): 3056. Дои:10.1182 / кровь-2014-04-567289.
- ^ Hantschel, O (2015). «Нацеливание на BCR-ABL и JAK2 во всех Ph ALL». Кровь. 122 (13): 2167–2175. Дои:10.1182 / кровь-2014-12-617548. PMID 25721043.
- ^ Bandyopadhyay, G (2004). «Хлорогеновая кислота ингибирует тирозинкиназу Bcr-Abl и запускает зависимый от митоген-активируемой протеинкиназы p38 апоптоз в клетках хронического миелолейкоза». Кровь. 104 (8): 2514–2522. Дои:10.1182 / кровь-2003-11-4065. PMID 15226183.
- ^ Скорски, Т. (1994). «Отрицательная регуляция активности p120GAP GTPase, стимулирующей активность p210bcr / abl: значение для RAS-зависимого роста положительных клеток по филадельфийской хромосоме». Журнал экспериментальной медицины. 179 (6): 1855–1865. Дои:10.1084 / jem.179.6.1855. ЧВК 2191514. PMID 8195713.
- ^ Стилман, Л.С.; Pohnert, S.C .; Shelton, J.G .; Франклин, Р. А .; Bertrand, F.E .; Маккубри, Дж. А. (2004). «JAK / STAT, Raf / MEK / ERK, PI3K / Akt и BCR-ABL в прогрессии клеточного цикла и лейкемогенезе». Лейкемия. 18 (2): 189–218. Дои:10.1038 / sj.leu.2403241. PMID 14737178.
- ^ Burke, B.A .; Кэрролл, М. (2010). «BCR – ABL: многогранный промотор мутации ДНК при хроническом миелогенном лейкозе». Лейкемия. 24 (6): 1105–1112. Дои:10.1038 / leu.2010.67. ЧВК 4425294. PMID 20445577.
- ^ «Тирозинкиназа c-Abl отвечает на повреждение ДНК путем активации гомеодомен-взаимодействующей протеинкиназы 2». Журнал биологической химии. 290 (27): 16489. 2015. Дои:10.1074 / jbc.p114.628982. ЧВК 4505403.
- ^ Kang, Z .; Liu, Y .; Xu, L .; Long, Z .; Huang, D .; Ян, Ю. (2011). «Филадельфийская хромосома». Ссылка Springer. Дои:10.1007 / springerreference_103128.
- ^ Кипреос, E.T .; Ван, J.Y. (1992). «Клеточный цикл - регулируемое связывание тирозинкиназы c-Abl с ДНК». Наука. 256 (3055): 382–385. Bibcode:1992Наука ... 256..382K. Дои:10.1126 / science.256.5055.382. PMID 1566087.
- ^ Qazi, S .; Укун, Ф. (2013). «Частота и биологическое значение делеций гена IKZF1 / Ikaros в педиатрической филадельфийской хромосоме отрицательной и филадельфийской хромосоме положительной B-клеточной предшественнице острого лимфобластного лейкоза». Haematologica. 98 (12): e151 – e152. Дои:10.3324 / haematol.2013.091140. ЧВК 3856976. PMID 24323986.
- ^ Шиндлер Т., Борнманн В., Пеллицена П., Миллер В. Т., Кларксон Б., Куриян Дж. (2000). «Структурный механизм ингибирования STI-571 тирозинкиназы Абельсона». Наука. 289 (5486): 1857–9. Bibcode:2000Sci ... 289.1938S. Дои:10.1126 / science.289.5486.1938. PMID 10988075.
- ^ Nimmanapalli, R .; Бхалла, К. (2002). «Новые таргетные методы лечения острых лейкозов, положительных по Bcr – Abl: помимо STI571». Онкоген. 21 (56): 8584–8590. Дои:10.1038 / sj.onc.1206086. PMID 12476305.
- ^ Дэн, С .; Naito, M .; Цуруо, Т. (1998). «Избирательная индукция апоптоза в филадельфийских хромосомно-положительных клетках хронического миелогенного лейкоза ингибитором тирозинкиназы BCR-ABL, CGP 57148». Гибель клеток и дифференциация. 5 (8): 710–715. Дои:10.1038 / sj.cdd.4400400. PMID 10200527.
- ^ Чай Пемовская; Эрик Джонсон; Мика Контро; Гретхен А. Репаски; Джеффри Чен; Питер Уэллс; Чиаран Н. Кронин; Мишель Мактигу; Олли Каллиониеми; Киммо Поркка; Брайон В. Мюррей; Кристер Веннерберг (март 2015 г.). «Акситиниб эффективно ингибирует BCR-ABL1 (T315I) с четко выраженной конформацией связывания». Природа. 519 (7541): 102–105. Bibcode:2015Натура.519..102П. Дои:10.1038 / природа14119. PMID 25686603.
- ^ Онкологический центр Fox Chase (2015-12-03). «50 лет со дня открытия филадельфийской хромосомы». Цитировать журнал требует
| журнал =(помощь) - ^ Новелл П., Хангерфорд Д. (ноябрь 1960 г.). «Минутная хромосома при хроническом гранулоцитарном лейкозе». Наука. 132 (3438): 1488–1501. Bibcode:1960Sci ... 132.1488.. Дои:10.1126 / science.132.3438.1488. PMID 17739576.
- ^ Роули Дж. Д. (1973). «Письмо: новая стойкая хромосомная аномалия при хроническом миелогенном лейкозе, идентифицированная с помощью флюоресценции хинакрина и окрашивания по Гимзе». Природа. 243 (5405): 290–3. Bibcode:1973Натура.243..290р. Дои:10.1038 / 243290a0. PMID 4126434.
- ^ Клаудиа Дрейфус (2011-02-07). «Матриарх современной генетики рака». Нью-Йорк Таймс.
внешняя ссылка
| Классификация |
|---|
- Филадельфия + хромосома в Национальной медицинской библиотеке США Рубрики медицинской тематики (MeSH)
- bcr-abl + Fusion + белки в Национальной медицинской библиотеке США Рубрики медицинской тематики (MeSH)