Общий синтез озельтамивира - Oseltamivir total synthesis - Wikipedia

Общий синтез озельтамивира касается полный синтез препарата против гриппа осельтамивир[1] проданный Hoffmann-La Roche под торговое наименование Тамифлю. Его коммерческое производство начинается с биомолекула шикимовая кислота собрано из Китая звездчатый анис с ограниченными поставками по всему миру. Из-за его ограниченного предложения ведутся поиски альтернативных путей синтеза, предпочтительно не требующих шикимовой кислоты, и на сегодняшний день опубликовано несколько таких путей. Важен контроль стереохимии: в молекуле три стереоцентры и искомый изомер представляет собой только 1 из 8 стереоизомеров.
Коммерческое производство
Текущий метод производства основан на первом масштабируемом синтезе, разработанном Gilead Sciences[2] начиная с встречающихся в природе хинная кислота или же шикимовая кислота. Из-за более низких выходов и требуемых дополнительных стадий (из-за дополнительной дегидратации) путь хинной кислоты был отклонен в пользу способа на основе шикимовой кислоты, который получил дальнейшие улучшения благодаря Hoffmann-La Roche.[3][4]Текущий промышленный синтез резюмируется ниже:

Синтез Карпфа / Труссарди
Текущий метод производства включает две стадии реакции с потенциально опасными азиды. Сообщенный синтез тамифлю без азидов представлен компанией Roche графически:[5]
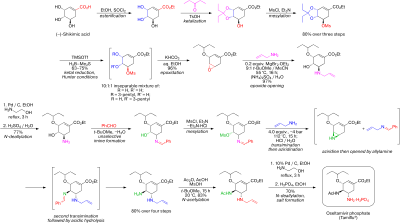
Синтез начинается с естественно доступных (-) -шикимовая кислота. 3,4-пентилиденацеталь мезилат готовится в три этапа: этерификация с этиловый спирт и тионилхлорид; кетализация с п-толуолсульфоновая кислота и 3-пентанон; и мезилирование с триэтиламин и метансульфонилхлорид. Редукционное открытие кеталь в модифицированных условиях Хантера[6] в дихлорметан дает неразрывную смесь изомерный мезилаты. Соответствующие эпоксид формируется в базовых условиях с бикарбонат калия. Используя недорогой Кислота Льюиса диэтилэфират бромида магния (обычно готовится свежим путем добавления магний обращается к 1,2-дибромэтан в бензол:диэтиловый эфир ) эпоксид открывается с аллиламин с получением соответствующего 1,2-аминоспирта. Несмешивающийся с водой растворители метил-трет-бутиловый эфир и ацетонитрил используются для упрощения процедуры обработки, которая включает перемешивание с 1 М водным сульфат аммония. Сокращение на палладий, продвигаемый этаноламин с последующей кислотной обработкой получали 1,2-аминоспирт со снятой защитой. Аминоспирт был преобразован непосредственно в соответствующий аллилдиамин в интересной каскадной последовательности, которая начинается с неселективной воображение из бензальдегид с азеотропный удаление воды метил-трет-бутиловым эфиром. Мезилирование с последующим удалением твердого побочного продукта триэтиламин гидрохлорид, приводит к промежуточному продукту, который должен был пройти азиридирование на трансмиссия с другим эквивалентом аллиламина. С либрированным метансульфоновая кислота, тоазиридин полностью открывается, давая диамин, который немедленно подвергается второму трансмилицированию. Кислая гидролиз затем удалил я добываю. Селективный ацилирование с уксусный ангидрид (под буферизованный условиях 5-аминогруппа протонированный из-за значительной разницы в пKа, 4,2 против 7,9, предотвращение ацетилирование ) дает желаемый N-ацетилированный продукт в кристаллической форме после экстракционной обработки. Ну наконец то, деаллиляция как указано выше, дал свободная база осельтамивира, который был преобразован в желаемый фосфат осельтамивира путем обработки фосфорная кислота. Конечный продукт получают с высокой чистотой (99,7%) и общим выходом 17-22% по (-) - шикимовой кислоте. Отмечено, что при синтезе исключается использование потенциально взрывоопасных азид реагенты и промежуточные продукты; однако в синтезе, фактически используемом Roche, используются азиды. У компании Рош есть другие способы использования сельтамивира, которые не включают использование (-) - шикимовой кислоты в качестве исходного материала хирального пула, например, путь Дильса-Альдера, включающий фуран и этилакрилат или изофталевая кислота путь, который включает каталитическое гидрирование и ферментативную десимметризацию.
Кори синтез
В 2006 году группа E.J. Кори опубликовали новый способ обхода шикимовой кислоты, начиная с бутадиен и акриловая кислота.[7] Изобретатели предпочли не патент эта процедура описана ниже.

Бутадиен 1 реагирует в асимметричный Реакция Дильса-Альдера с этерификация продукт акриловая кислота и 2,2,2-трифторэтанол 2 катализируется Катализатор CBS. В сложный эфир 3 превращается в амид в 4 по реакции с аммиак и следующий шаг к лактам 5 является йодолактамизация с йод по инициативе триметилсилилтрифлат. Амидная группа снабжена BOC защитная группа по реакции с Boc ангидрид в 6 и йодный заместитель удаляется в реакция элиминации с DBU к алкен 7. Бром вводится в 8 аллильным бромированием с NBS и амидная группа расщепляется этиловый спирт и карбонат цезия сопровождается отщеплением бромида до диенэтилового эфира 9. Вновь образованная двойная связь функционализирована N-бромацетамид 10 катализируетсябромид олова (IV) с полным контролем стереохимия. На следующем этапе атом брома в 11 является перемещенный атомом азота в амидной группе с сильным основанием ХМДС к азиридин 12 который, в свою очередь, открывается реакцией с 3-пентанолом 13 к эфир 14. На последнем этапе группа BOC удаляется с помощью фосфорная кислота и фосфат осельтамивира 15 сформирован.
Синтез Шибасаки
Также в 2006 году группа Масакацу Шибасаки из Токийский университет опубликовали синтез снова в обход шикимовой кислоты.[8][9]


Улучшенный метод, опубликованный в 2007 году, начинается с энантиоселективный десимметризация из азиридин 1 с триметилсилилазид (TMSN3) и хиральный катализатор к азид 2. В амид группа защищена как группа BOC с Boc ангидрид и DMAP в 3 и йодолактамизация с йод и карбонат калия сначала дает нестабильное промежуточное звено 4 а затем устойчивый циклический карбамат 5 после устранение из йодистый водород с DBU.
Амидная группа защищена как BOC 6 и азидная группа превращается в амид 7 восстановительным ацилированием тиоуксусная кислота и 2,6-лутидин. Карбонат цезия выполняет гидролиз карбаматной группы к алкоголь 8 который впоследствии окисляется до кетон 9 с Десс-Мартин периодинан. Цианофосфорилирование с диэтилфосфороцианидат (DEPC) изменяет кетонную группу на цианофосфат 10 прокладывая путь для внутримолекулярный аллильная перегруппировка к нестабильному β-аллилу фосфат 11 (толуол, герметичная пробирка), гидролизованный до спирта 12 с хлорид аммония. Эта гидроксильная группа имеет неправильную стереохимию и поэтому перевернутый в Мицунобу реакция с п-нитробензойная кислота с последующим гидролизом п-нитробензоата до 13.
Вторая реакция Мицунобу затем формирует азиридин 14 доступен для реакции раскрытия кольца с 3-пентанол катализируется трифторид бора в эфир 15. На последнем этапе удаляется группа BOC (HCl) и фосфорная кислота добавлен к цели 16.
Синтез Фукуямы
Подход, опубликованный в 2007 г.[10] как у Кори начинается с асимметричная реакция Дильса-Альдера на этот раз с исходными материалами пиридин и акролеин.


Пиридин (1) является уменьшенный с борогидрид натрия в присутствии бензил хлорформиат к КБЗ защищенный дигидропиридин 2. Асимметричная реакция Дильса-Альдера с акролеин 3 осуществляется с Катализатор Макмиллана к альдегид 4 как эндо-изомер который окисляется до карбоновая кислота 5 с хлорит натрия, монокалиевый фосфат и 2-метил-2-бутен. Добавление бром дает галолактонизация товар 6 и после замены защитной группы Cbz на BOC защитная группа в 7 (гидрогенолиз в присутствии ди-терт-бутилдикарбонат ) а карбонил группа вводится в промежуточном 8 каталитическим оксид рутения (IV) и жертвенный катализатор периодат натрия. Добавление аммиак расщепляет сложноэфирную группу с образованием амид 9 то алкоголь группа из которых мезилированный соединять 10. На следующем этапе иодобензолдиацетат добавляется, превращая амид в Перегруппировка Гофмана аллилу карбамат 12 после улавливания промежуточного изоцианата аллиловый спирт 11. При добавлении этоксид натрия в этаноле одновременно протекают три реакции: расщепление амид образовать новый этил сложный эфир группа, замещение мезильной группы новообразованным ВОС защищенным амин для азиридин группа и реакция элиминации формирование алкен группа в 13 с выделением HBr. На последних двух этапах азиридиновое кольцо открывается посредством 3-пентанол 14 и трифторид бора к аминоэфиру 15 с заменой группы BOC на ацил группы и при удалении другой защитной группы амина (Pd / C, Ph3п, и 1,3-диметилбарбитуровая кислота в этаноле) и добавление фосфорная кислота осельтамивир 16 получается.
Синтез Троста

В 2008 году группа Барри М. Трост из Стэндфордский Университет опубликовал самый короткий синтетический маршрут на сегодняшний день.[11]
Синтез Хаяши
В 2009 году Hayashi et al. успешно разработал эффективный и недорогой синтетический способ получения (-) - осельтамивира (1). Их цель заключалась в разработке процедуры, подходящей для крупномасштабного производства. Принимая во внимание стоимость, доходность и количество синтетических шагов, энантиоселективный полный синтез из (1) было выполнено с помощью трех операций с одним горшком.[12][3] Использование Хаяши и др. Операций с одним резервуаром позволило им выполнить несколько этапов реакции в одном резервуаре, что в конечном итоге минимизировало количество необходимых этапов очистки, потери и сэкономило время.

Во-первых операция с одним горшком, Hayashi et al. начинается с использования силилового эфира дифенилпролинола (4)[4] как органокатализатор, наряду с алкоксиальдегидом (2) и нитроалкен (3) выполнить асимметричный Реакция Майкла, предоставляя энантиоселективный Майкл аддукт. При добавлении производного диэтилвинилфосфата (5) Михаилу аддукт, домино Реакция Майкла и Хорнер-Уодсворт-Эммонс реакция происходит из-за фосфонатной группы, полученной из (5) с получением производного этилциклогексенкарбоксилата вместе с двумя нежелательными побочными продуктами. Чтобы превратить нежелательные побочные продукты в желаемое производное этилциклогексенкарбоксилата, смесь продукта и побочных продуктов обрабатывали Cs2CO3 в этаноле. Это вызвало ретро-реакцию Михаэля на один побочный продукт и ретро-реакцию.альдольная реакция сопровождается реакцией Хорнера-Уодсворта-Эммонса на другого. Оба побочных продукта были успешно преобразованы в желаемые производные. Наконец, добавление п-толуентиол с Cs2CO3 дает (6) с выходом 70% после очистки колоночная хроматография, с преобладанием желаемого изомера.[12]

В секунду операция с одним горшком, трифторуксусная кислота сначала используется для снятия защиты терт-бутиловый эфир (6); любой избыток реагента удаляли упариванием. Карбоновая кислота, полученная в результате снятия защиты, затем превращалась в ацилхлорид путем оксалилхлорид и каталитическое количество DMF. Наконец, добавление азида натрия в последней реакции второго реактора в одной емкости дает ацилазид (7) без какой-либо очистки.[12]

Финал операция с одним горшком начинается с Курций перестановка ацилазида (7) с образованием изоцианатной функциональной группы при комнатной температуре. В изоцианат производная затем реагирует с уксусная кислота с получением желаемой ацетиламиногруппы, обнаруженной в (1). Эта перегруппировка домино Курциуса и образование амида происходит в отсутствие тепла, что чрезвычайно полезно для уменьшения любой возможной опасности. Нитро-фрагмент (7) восстанавливается до желаемого амина, наблюдаемого в (1) с Zn / HCl. Из-за суровых условий нитро-восстановления для нейтрализации реакции использовали аммиак. Карбонат калия затем был добавлен, чтобы дать (1) через ретро-реакцию Михаэля тиол. (1) затем очищали кислотно-щелочной экстракцией. Общий выход для полного синтеза (-) - осельтамивира составляет 57%.[12] Hayashi et al. Использование недорогих, безопасных реагентов позволило создать эффективный, высокоэффективный синтетический путь, который может позволить производить огромное количество новых производных в надежде на борьбу с вирусами, устойчивыми к (-) - осельтамивиру.
Рекомендации
- ^ Классика полного синтеза III: дальнейшие цели, стратегии, методы К. К. Николау, Джейсон С. Чен ISBN 978-3-527-32957-1 2011
- ^ Rohloff John C .; Kent Kenneth M .; Постич Майкл Дж .; Беккер Марк В .; Chapman Harlan H .; Келли Дафни Э .; Лью Уиллард; Луи Майкл С .; McGee Lawrence R .; и другие. (1998). «Практический полный синтез лекарственного средства против гриппа GS-4104». J. Org. Chem. 63 (13): 4545–4550. Дои:10.1021 / jo980330q.
- ^ а б Лаборда, Педро; Ван Су-Янь; Фогльмейр, Йозеф (11 ноября 2016 г.). «Ингибиторы нейраминидазы гриппа: синтетические подходы, производные и биологическая активность». Молекулы. 21 (11): 1513. Дои:10.3390 / молекулы21111513. ЧВК 6274581. PMID 27845731.
- ^ а б Хаяси, Юдзиро; Гото, Хироаки; Хаяси, Такааки; Сёдзи, Мицуру (2004-07-04). «Силиловые эфиры дифенилпролинола как эффективные органокатализаторы для асимметричной реакции Михаэля альдегидов и нитроалкенов». Angewandte Chemie International Edition. 44 (27): 4212–4215. Дои:10.1002 / anie.200500599. ISSN 1521-3773. PMID 15929151.
- ^ Карпф, М; Труссарди, Р. (март 2001 г.). «Новое безазидное превращение эпоксидов в 1,2-диаминосоединения: синтез противогриппозного ингибитора нейраминидазы осельтамивир фосфата (Тамифлю)». J. Org. Chem. 66: 2044–51. Дои:10.1021 / jo005702l. PMID 11300898..
- ^ Биргит Бартельс; Роджер Хантер (1993). «Исследование селективности восстановления активированного кеталя с борандиметилсульфидом». J. Org. Chem. 58 (24): 6756–6765. Дои:10.1021 / jo00076a041.
- ^ Юнг, Инь-Юнг; Хонг, Сону; Кори, Э. Дж. (2006). «Короткий энантиоселективный путь синтеза антигриппозного ингибитора нейрамидазы осельтамивира из 1,3-бутадиена и акриловой кислоты». Варенье. Chem. Soc. 128 (19): 6310–6311. Дои:10.1021 / ja0616433. PMID 16683783.
- ^ Фукута, Юхей (2006). «Синтез De Novo Тамифлю посредством каталитического асимметричного раскрытия кольца мезо-азиридинов с TMSN 3». Журнал Американского химического общества. 128: 6312–6313. Дои:10.1021 / ja061696k. PMID 16683784.
- ^ Мита, Цуёси (2007). "Каталитический асимметричный синтез второго поколения Тамифлю: путь замещения аллила". Органические буквы. 9: 259–262. Дои:10.1021 / ol062663c.
- ^ Сато, Нобухиро (2007). «Практический синтез (-) - осельтамивира». Angewandte Chemie International Edition. 46: 5734–5736. Дои:10.1002 / anie.200701754.
- ^ Трост, Барри М. (2008). «Краткий синтез (-) - осельтамивира». Angewandte Chemie International Edition. 47: 3759–3761. Дои:10.1002 / anie.200800282.
- ^ а б c d Исикава, Хаято; Сузуки, Такаки; Хаяси, Юдзиро (02.02.2009). «Высокоурожайный синтез ингибитора нейрамидазы против гриппа (-) - осельтамивира с помощью трех операций в одной банке». Angewandte Chemie International Edition. 48 (7): 1304–1307. Дои:10.1002 / anie.200804883. ISSN 1521-3773. PMID 19123206.