Селенорганическая химия - Organoselenium chemistry
Селенорганические соединения (или селеноорганические) являются химические соединения содержащий углерод -к-селен химические связи. Селенорганическая химия это соответствующая наука, изучающая их свойства и реакционную способность.[1][2][3][4] Селен вместе с кислородом и серой относится к группа 16 элементов или халькогены, и сходства в химии следовало ожидать.
Селен может существовать с степень окисления −2, +2, +4, +6. Se (II) является доминирующей формой в химии селенорганических соединений. В столбце 16 группы прочность сцепления становится все слабее (234 кДж /моль для связи C-Se и 272 кДж / моль для связи C-S) и длина облигаций длиннее (C-Se 198 pm, C-S 181 pm и C-O 141 pm). Соединения селена больше нуклеофильный чем соответствующие соединения серы, а также более кислые. В пKа значения XH2 равны 16 для кислорода, 7 для серы и 3,8 для селена. В отличие от сульфоксиды соответствующие селеноксиды нестабильны в присутствии β-протонов, и это свойство используется во многих органические реакции селена, особенно при окислении селеноксида и при удалении селеноксида. Селенорганические соединения обнаруживаются в следовых количествах в окружающих водах, почвах и отложениях.[5]
Первым выделенным селенорганическим соединением было диэтилселенид в 1836 г.[6][7]
Структурная классификация селенорганических соединений

- Селенолы (RSeH) - селеновые эквиваленты спирты и тиолы. Эти соединения относительно нестабильны и обычно имеют неприятный запах. Бензолезеленол (также называемый селенафенолом или PhSeH) более кислый (pKа 5.9), чем тиофенол (pKа 6.5), а также легче окисляется до диселенид. Селенафенол получают путем восстановления дифенилдиселенида.[8]
- Диселениды (R − Se − Se − R) - селеновые эквиваленты перекиси и дисульфиды. Они являются полезными и стабильными при хранении предшественниками более реакционноспособных селеносодержащих реагентов, таких как селенолы и селанилгалогениды. Наиболее известен в органической химии дифенилдиселенид, приготовленный из фенилмагний бромид и селен с последующим окислением продукта PhSeMgBr.[9]
- Галогениды селанила (R-Se-Cl, R-Se-Br) получают галогенированием диселенидов. Бромирование дифенилдиселенида дает фенилселанилбромид (PhSeBr). Эти соединения являются источниками «PhSe+".
- Селениды (R − Se − R), также называемый селеноэфиры, являются селеновыми эквивалентами эфиры и сульфиды. Одним из примеров является диметилселенид ((CH3)2Se). Это наиболее распространенные селенорганические соединения. Симметричные селениды обычно получают алкилированием солей селенидов щелочных металлов, например селенид натрия. Несимметричные селениды получают алкилированием селеноатов. Эти соединения обычно реагируют как нуклеофилы, например с алкилгалогениды (R'− X), чтобы дать соли селенония R'RRSe+Икс−. Двухвалентный селен также может взаимодействовать с мягкими гетероатомами с образованием центров гипервалентного селена.[7] В некоторых случаях они также реагируют как электрофилы, например с литийорганический реагенты (R'Li) к ел комплекс R'RRSe−Ли+.
- Селеноксиды (R − Se (O) −R) - селеновые эквиваленты сульфоксиды. В дальнейшем они могут быть окислены до селеноны R − Se (O)2R, селеновые аналоги сульфоны.
- SeO-Селенопероксолы (RSe-OH; ранее селененовые кислоты) являются промежуточными продуктами окисления селенолов. Они встречаются в некоторых селеноферментах, таких как глутатионпероксидаза.
- Селениновые кислоты (RSe (O) OH) являются аналогами сульфиновые кислоты.
- Пероксиселениновые кислоты (RSe (O) OOH) катализировать эпоксидирование реакции и Окисления Байера-Виллигера.
- Селенураны находятся гипервалентный селенорганические соединения, формально полученные из тетрагалогенидов, таких как SeCl4. Примеры типа ArSeCl3.[10] Хлориды получают хлорированием селененилхлорид.
- Селениран трехчленные кольца (родитель: C2ЧАС4Se) связанные с тиираны но, в отличие от тииранов, селенираны кинетически нестабильны, экструдируя селен напрямую (без окисления) с образованием алкены. Это свойство было использовано в синтетической органической химии.[11]
- Селонес (Р2C = Se) - селеновые аналоги кетонов. Они редки из-за их склонности к олигомеризовать.[12] Диселенобензохинон стабилен как комплекс металла.[13] Селеномочевина представляет собой пример стабильного соединения, содержащего связь C = Se.
- Селенотиопероксиды (R − Se − S − R), соединения со связями селен – сера, аналогичные дисульфиды.
Селенорганические соединения в природе
Селен в форме селенорганических соединений является важным микронутриентом, отсутствие которого в рационе вызывает дисфункцию сердечной мышцы и скелета. Селенорганические соединения необходимы для защиты клеток от окислительного повреждения и для правильного функционирования иммунной системы. Они также могут играть роль в предотвращении преждевременного старения и рака. Источником селена, используемого в биосинтезе, является селенофосфат.
Глутатионоксидаза представляет собой фермент, в активном центре которого находится селенол. Селенорганические соединения обнаружены у высших растений. Например, при анализе чеснока по методике высокоэффективная жидкостная хроматография в сочетании с масс-спектрометрия с индуктивно связанной плазмой (ВЭЖХ-ИСП-МС) было обнаружено, что γ-глутамил-Se-метилселеноцистеин был основным Se-содержащим компонентом, наряду с меньшими количествами Se-метилселеноцистеин. Следовые количества диметилселенид и аллилметилселенид обнаруживаются в дыхании человека после употребления сырого чеснока.[14]
Селеноцистеин и селенометионин
Селеноцистеин, называемая двадцать первой аминокислотой, необходима для управляемого рибосомами синтеза белка в некоторых организмах.[15] В настоящее время известно более 25 селенсодержащих белков (селенопротеинов).[16] Большинство селензависимых ферментов содержат селеноцистеин, что связано с цистеин аналог, но с селеном вместо серы. Этот аминокислота является закодированный особым образом с помощью ДНК.
Селенометионин представляет собой селенидсодержащую аминокислоту, которая также встречается в природе, но образуется в результате посттранскрипционной модификации.
Селенорганическая химия в органическом синтезе
Селенорганические соединения представляют собой специализированный, но полезный набор реагентов, используемых в органическом синтезе, хотя они обычно исключаются из процессов, используемых в фармацевтике, из-за проблем с нормативным регулированием. Их полезность зависит от определенных атрибутов, включая (i) слабость связи C-Se и (ii) легкое окисление соединений двухвалентного селена.
Виниловые селениды
Виниловые селениды представляют собой селенорганические соединения, которые играют роль в органическом синтезе, особенно в разработке удобных стереоселективный пути к функционализированным алкены.[17] Хотя для получения виниловых селенидов упоминаются различные методы, более полезная процедура была сосредоточена на нуклеофильный или же электрофильный селенорганическое присоединение к концевым или внутренним алкины.[18][19][20][21] Например, нуклеофильное присоединение превращение селенофенола в алкины дает, предпочтительно, Z-виниловые селениды после более продолжительного времени реакции при комнатной температуре. Реакция идет быстрее при высокой температуре; однако смесь Z- и E-виниловых селенидов была получена в соотношении почти 1: 1.[22] С другой стороны, аддукты зависят от природы заместители на тройная связь. И наоборот, виниловые селениды могут быть получены палладий -катализируемое гидроселенирование алкины давать марковниковский аддукт с хорошими выходами. Существуют некоторые ограничения, связанные с методиками получения виниловых селенидов, показанными выше; описанные процедуры используют диорганоилдиселениды или селенофенол в качестве исходных материалов, которые являются летучими и нестабильными и имеют неприятный запах. Кроме того, получение этих соединений сложное.
Селеноксидные окисления
Диоксид селена полезно в органическое окисление. В частности, SeO2 преобразует аллильный метиленовая группа в соответствующий алкоголь. Эту реакцию вызывает ряд других реагентов.

С точки зрения механизм реакции, SeO2 и аллильный субстрат реагируют через перициклический процесс, начинающийся с ее реакция который активирует связь C-H. Второй шаг - это [2,3] сигматропная реакция. Окисление с участием диоксида селена часто проводят с каталитическими количествами соединения селена и в присутствии жертвенный катализатор или сооксидант, такой как пероксид водорода.
SeO2-основные окисления иногда дают карбонильные соединения, такие как кетоны,[23] β-Пинен[24] и циклогексанон окисление до 1,2-циклогександиона.[25] Окисление кетоны с α-метиленовыми группами дает дикетоны. Этот тип окисления оксидом селена называется Окисление Райли.[26]
Удаление селеноксида
В присутствии β-водорода селенид дает реакция элиминации после окисления оставить после себя алкен и SeO-селенопероксол. В SeO-селенопероксол обладает высокой реакционной способностью и не выделяется как таковой. В реакции элиминирования все пять участвующих реакционных центров являются копланарный и, следовательно, стереохимия реакции син. Используемые окислители: пероксид водорода, озон или же MCPBA. Этот тип реакции часто используется с кетоны ведущий к Enones. Примером может служить отщепление ацетилциклогексанона с помощью бензолселенилхлорид и гидрид натрия.[27]
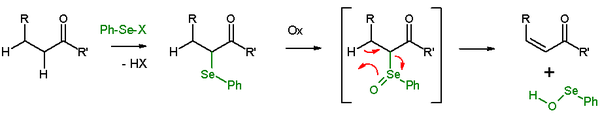
В Устранение Грико аналогичное элиминирование селеноксида с использованием о-нитрофенилселеноцианата и трибутилфосфина, чтобы вызвать элиминацию элементов H2О.
Функционализация олефинов
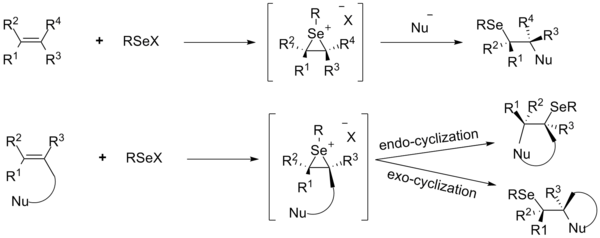
За последние два десятилетия[когда? ] Особое внимание уделяется функционализации двойных углерод-углеродных связей присоединением электрофильных селенорганических соединений. Реакция селанилирования инициируется образованием промежуточных соединений иона селенирана из алкенов и селенового электрофила RSeX с последующей нуклеофильной атакой на обратной стороне, приводящей к продукту анти-присоединения. Атака нуклеофила происходит на атоме углерода, который имеет более стабильный положительный заряд, обычно это наиболее замещенный атом углерода. Реакции присоединения различных электрофилов селена к алкенам были подробно изучены с использованием внутренних и внешних нуклеофилов.[28]
Рекомендации
- ^ А. Криф, Л. Хевеси, Селеноорганическая химия I. Превращения функциональных групп.., Шпрингер, Берлин, 1988 ISBN 3-540-18629-8
- ^ С. Патай, З. Раппопорт (ред.), Химия органических соединений селена и теллура., Джон. Wiley and Sons, Чичестер, Vol. 1, 1986 ISBN 0-471-90425-2
- ^ Паульмье, К. Селеновые реагенты и промежуточные продукты в органическом синтезе; Baldwin, J. E., Ed .; Pergamon Books Ltd.: Нью-Йорк, 1986 ISBN 0-08-032484-3
- ^ Freudendahl, Diana M .; Санторо, Стефано; Шахзад, Сохаил А .; Санти, Клаудио; Вирт, Томас (2009). «Зеленая химия с селеновыми реагентами: разработка эффективных каталитических реакций». Angewandte Chemie International Edition. 48 (45): 8409–11. Дои:10.1002 / anie.200903893. PMID 19802863.
- ^ Wallschläger, D .; Фельдманн, Ф. (2010). Образование, наличие, значение и анализ селенорганических и целлюлозноорганических соединений в окружающей среде. Ионы металлов в науках о жизни. 7, Металлоорганические соединения в окружающей среде и токсикологии. Издательство РСК. С. 319–364. ISBN 978-1-84755-177-1.
- ^ Лёвиг, К. Дж. (1836). "Ueber schwefelwasserstoff — und selenwasserstoffäther" [О сероводороде и водородном эфире селена]. Annalen der Physik. 37: 550–553.
- ^ а б Мукерджи, Анна Дж .; Zade, Sanjio S .; Singh, Harkesh B .; Сунодж, Рагхаван Б. (2010). «Селенорганическая химия: роль внутримолекулярных взаимодействий». Химические обзоры. 110 (7): 4357–4416. Дои:10.1021 / cr900352j. PMID 20384363.
- ^ Органический синтез, Сб. Vol. 3, стр. 771 (1955); Vol. 24, стр. 89 (1944) Интернет-статья.
- ^ Органический синтез, Сб. Vol. 6, стр. 533 (1988); Vol. 59, стр. 141 (1979) Статья
- ^ Химия гипервалентных соединений (1999) Кин-я Акиба ISBN 978-0-471-24019-8
- ^ Связь Разработки химии селенагетероциклических соединений, имеющие практическое значение для синтеза и медицинской биологии. Аркивок 2006 (JE-1901MR) Яцек Млоховски, Кристиан Клок, Рафал Лисиак, Петр Потачек и Галина Войтович
- ^ Окадзаки, Р .; Токито, Н. (2000). «Тяжелые кетоны, более тяжелые конгенеры элементов кетона». Отчеты о химических исследованиях. 33 (9): 625–630. Дои:10.1021 / ar980073b. PMID 10995200.
- ^ Amouri, H .; Moussa, J .; Ренфрю, А. К .; Дайсон, П. Дж .; Rager, M. N .; Шаморо, Л.-М. (2010). «Открытие, структура и противораковая активность иридиевого комплекса дизеленобензохинона». Angewandte Chemie International Edition. 49 (41): 7530–7533. Дои:10.1002 / anie.201002532. PMID 20602399.
- ^ Блок, Э. (2010). Чеснок и другие луковые культуры: знания и наука. Королевское химическое общество. ISBN 978-0-85404-190-9.
- ^ Axley, M.J .; Böck, A .; Штадтман, Т. (1991). «Каталитические свойства кишечная палочка мутант формиатдегидрогеназы, в котором сера заменяет селен ». Proc. Natl. Акад. Sci. СОЕДИНЕННЫЕ ШТАТЫ АМЕРИКИ. 88 (19): 8450–8454. Bibcode:1991PNAS ... 88.8450A. Дои:10.1073 / пнас.88.19.8450. ЧВК 52526. PMID 1924303.
- ^ Papp, L.V .; Lu, J .; Holmgren, A .; Ханна, К. (2007). «От селена до селенопротеидов: синтез, идентичность и их роль в здоровье человека». Антиоксиданты и редокс-сигналы. 9 (7): 775–806. Дои:10.1089 / ars.2007.1528. PMID 17508906.
- ^ Комассето, Жоао Вальдир; Линг, Ло Вай; Петраньяни, Никола; Стефани, Хелио Александр (1997). «Виниловые селениды и теллуриды - получение, реакционная способность и синтетические применения». Синтез. 1997 (4): 373. Дои:10.1055 / с-1997-1210.
- ^ Комассето, Дж (1983). «Виниловые селениды». Журнал металлоорганической химии. 253 (2): 131–181. Дои:10.1016 / 0022-328X (83) 80118-1.
- ^ Зени, Гилсон; Stracke, Marcelo P .; Ногейра, Кристина В .; Брага, Антонио Л .; Menezes, Paulo H .; Стефани, Хелио А. (2004). «Гидроселенирование алкинов бутилселенолатом лития: подход к синтезу виниловых селенидов». Органические буквы. 6 (7): 1135–8. Дои:10.1021 / ol0498904. PMID 15040741.
- ^ Дабдоуб, М. (2001). «Синтез (Z) -1-фенилселено-1,4-диорганил-1-бутен-3-инов: гидроселенирование симметричных и несимметричных 1,4-диорганил-1,3-бутадиинов». Тетраэдр. 57 (20): 4271–4276. Дои:10.1016 / S0040-4020 (01) 00337-4.
- ^ Дорегобаррос, О; Lang, E; Деоливейра, К; Пеппе, С; Зени, Г. (2002). «Опосредованное йодидом индия (I) химио-, регио- и стереоселективное гидроселенирование производных 2-алкин-1-ола». Буквы Тетраэдра. 43 (44): 7921. Дои:10.1016 / S0040-4039 (02) 01904-4.
- ^ Комассето, Дж (1981). «Стереоселективный синтез виниловых селенидов». Журнал металлоорганической химии. 216 (3): 287–294. Дои:10.1016 / S0022-328X (00) 85812-X.
- ^ Органический синтез Coll. Vol. 9, стр. 396 (1998); Vol. 71, стр. 181 (1993) Интернет-статья В архиве 2005-10-24 на Wayback Machine
- ^ Органический синтез Coll. Vol. 6, стр. 946 (1988); Vol. 56, стр. 25 (1977). Интернет-статья В архиве 2005-11-01 на Wayback Machine
- ^ Органический синтез, Сб. Vol. 4, стр. 229 (1963); Vol. 32, стр. 35 (1952). Интернет-статья В архиве 2005-11-27 на Wayback Machine
- ^ Райли, Гарри Листер; Морли, Джон Фредерик; Друг, Норман Альфред Чайлд (1932). «255. Диоксид селена - новый окислитель. Часть I. Его реакция с альдегидами и кетонами». Журнал химического общества (возобновлено): 1875. Дои:10.1039 / JR9320001875.
- ^ Органический синтез Coll. Vol. 6, стр. 23 (1988); Vol. 59, стр. 58 (1979) Интернет-статья
- ^ Селеноорганическая химия: синтез и реакции - онлайн-библиотека Wiley. 2011. Дои:10.1002/9783527641949. ISBN 9783527641949.
Соединения углерод с другими элементами периодической таблицы | |
|---|---|
| Легенда |
|