Идиопатический фиброз легких - Idiopathic pulmonary fibrosis - Wikipedia
| Идиопатический фиброз легких | |
|---|---|
| Другие имена | Криптогенный фиброзирующий альвеолит, диффузный фиброзный альвеолит, обычный интерстициальный пневмонит[1] |
 | |
| Рисунок А показывает расположение легких и дыхательных путей в теле. На вставленном изображении показано подробное изображение дыхательных путей и воздушных мешков легких в разрезе. Рисунок B показывает фиброз (рубцевание) в легких. На вставке показано подробное изображение фиброза и того, как он повреждает дыхательные пути и воздушные мешочки.[1] | |
| Специальность | Пульмонология |
| Симптомы | Одышка, сухой кашель[1] |
| Осложнения | Легочная гипертония, сердечная недостаточность, пневмония, легочная эмболия[1] |
| Обычное начало | Постепенный[1] |
| Причины | Неизвестный[2] |
| Факторы риска | Курение сигарет, определенный вирусные инфекции, история семьи[1] |
| Диагностический метод | компьютерная томография, биопсия легкого[3] |
| Дифференциальная диагностика | Саркоидоз, Другой интерстициальные заболевания легких, гиперчувствительный пневмонит[4] |
| Уход | Легочная реабилитация, дополнительный кислород, трансплантация легких[1] |
| Медикамент | Пирфенидон, нинтеданиб[2] |
| Прогноз | Продолжительность жизни ~ 4 года[1] |
| Частота | 12 на 100 000 человек в год[4] |
Идиопатический фиброз легких (IPF) является типом хронического рубцевание заболевание легких характеризуется прогрессирующим и необратимым снижением легкое функция.[3][4] Ткань в легких становится толстой и жесткой, что влияет на ткань, окружающую воздушные мешочки в легких.[5] Симптомы обычно включают постепенное начало одышка и сухой кашель.[1] Другие изменения могут включать чувство усталости и аномально большие и куполообразные ногти на пальцах рук и ног (гвоздь).[1] Осложнения могут включать: легочная гипертония, сердечная недостаточность, пневмония, или же легочная эмболия.[1]
Причина неизвестна.[2] Факторы риска включают: курение сигарет, определенный вирусные инфекции и семейный анамнез заболевания.[1] Основной механизм включает рубцевание легких.[1] Диагностика требует исключения других потенциальных причин.[3] Это может быть поддержано компьютерная томография или же биопсия легкого которые показывают обычная интерстициальная пневмония (UIP).[3] Это тип интерстициальное заболевание легких (ILD).[3]
Люди часто получают выгоду от легочная реабилитация и дополнительный кислород.[1] Некоторые лекарства, такие как пирфенидон или же нинтеданиб может замедлить прогрессирование заболевания.[2] Трансплантация легких также может быть вариант.[1]
Во всем мире пострадали около 5 миллионов человек.[6] Заболевание впервые возникает примерно у 12 на 100 000 человек в год.[4] Чаще всего страдают люди в возрасте от 60 до 70 лет.[4] Мужчины болеют чаще, чем женщины.[4] Средний продолжительность жизни Следующий диагноз составляет около четырех лет.[1]
Признаки и симптомы


У многих людей симптомы присутствуют в течение значительного времени до постановки диагноза.[6] Наиболее частые клинические признаки IPF включают следующее:[3][7][8]
- Возраст старше 50 лет
- Сухой непродуктивный кашель при физической нагрузке
- Прогрессирующая одышка при физической нагрузке (одышка при физической нагрузке)
- Сухой инспираторный бибазиляр «на липучке» треск на аускультация (потрескивающий звук в легких во время вдоха, похожий на медленное отрывание липучки, слышен с помощью стетоскопа).[3][9][10]
- Клубок цифр, обезображивание кончиков пальцев рук или ног (см. изображение)
- Аномальный легочная функциональная проба результаты с признаками ограничения и нарушения газообмена.
Некоторые из этих особенностей связаны с хроническим гипоксемия (недостаток кислорода в крови), неспецифичны для IPF и могут возникать при других легочных заболеваниях. IPF следует рассматривать у всех пациентов с необъяснимой хронической одышкой при физической нагрузке, которые проявляются кашлем, бибазилярными хрипами на вдохе или удары пальцами.[3]
Оценка потрескивания «липучки» при аускультации легких - практический способ улучшить раннюю диагностику ИЛФ. Мелкие потрескивания легко распознаются клиницистами и характерны для IPF.[11]
Если двусторонние мелкие потрескивания присутствуют на протяжении всего времени вдоха и сохраняются после нескольких глубоких вдохов, и если они сохраняются несколько раз с интервалом в несколько недель у субъекта в возрасте ≥60 лет, это должно вызвать подозрение на IPF и привести к рассмотрению возможности проведения КТВР. сканирование грудной клетки более чувствительное, чем рентгенограмма грудной клетки.[10] Поскольку потрескивания не характерны для IPF, они должны требовать проведения тщательной диагностики.[3]
Причины
Причина IPF неизвестна, но было показано, что определенные факторы окружающей среды и воздействия повышают риск заражения IPF.[12] Курение сигарет является наиболее признанным и наиболее приемлемым фактором риска ИЛФ и увеличивает риск ИЛФ примерно в два раза.[12] Другие воздействия окружающей среды и производственного процесса, такие как воздействие металлической пыли, древесной пыли, угольной пыли, кремнезем, каменная пыль, биологическая пыль, образующаяся от сенной пыли или спор плесени или других сельскохозяйственных продуктов, а также профессии, связанные с сельским хозяйством / животноводством, также увеличивают риск ИЛЗ.[12] Есть некоторые свидетельства того, что вирусные инфекции могут быть связаны с идиопатическим фиброзом легких и другими заболеваниями. фиброзные заболевания легких.[13]
Патогенез
Несмотря на тщательное расследование, причина IPF остается неизвестной.[3] В фиброз в IPF был связан с курением сигарет, факторами окружающей среды (например, профессиональным воздействием газов, дыма, химикатов или пыли), другими заболеваниями, включая гастроэзофагеальная рефлюксная болезнь (ГЭРБ) или генетической предрасположенности (семейная IPF). Однако ни один из них не присутствует у всех людей с ИЛФ и, следовательно, не дает полностью удовлетворительного объяснения болезни.[3][14]
Считается, что IPF является результатом аберрантного процесса заживления ран, включая / включающий аномальное и чрезмерное отложение коллаген (фиброз) в легочной интерстиций с минимальными связанными воспаление.[15] Клеточное старение подозревается, что является основной причиной, и это мнение подтверждается преимуществами, наблюдаемыми у пациентов, принимающих сенолитический терапия.[16][17][18]
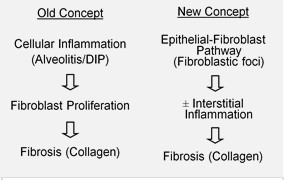
Предполагается, что первоначальное или повторяющееся повреждение при IPF происходит в клетках легких, называемых альвеолярными эпителиальными клетками (AECs, пневмоциты), которые выстилают большую часть альвеолярной поверхности.[19] Когда AEC типа I повреждены или потеряны, считается, что AEC типа II подвергаются разрастанию, чтобы покрыть открытые участки. подвальные мембраны. При нормальной репарации гиперпластические AEC типа II умирают, а оставшиеся клетки распространяются и подвергаются процессу дифференцировки, чтобы стать AEC типа I. При патологических условиях и при наличии трансформирующий фактор роста бета (TGF-β), фибробласты накапливаются в этих поврежденных областях и дифференцируются на миофибробласты которые выделяют коллаген и другие белки.[19] Раньше считалось, что воспаление был первым событием, вызвавшим рубцевание легочной ткани. Более поздние результаты показали, что развитие фибробластических очагов предшествует накоплению воспалительных клеток и последующему отложению коллагена.[20] Эта патогенетическая модель косвенно подтверждается клиническими особенностями IPF, включая скрытое начало в течение нескольких лет, относительно редкие острые обострения и отсутствие реакции на лечение. иммуносупрессивная терапия.[15][21] Ряд методов лечения, нацеленных на активацию фибробластов или синтез внеклеточного матрикса, в настоящее время проходят ранние испытания или рассматриваются для разработки.[нужна цитата ]
Семейная IPF составляет менее 5% от общего числа пациентов с IPF и клинически и гистологически неотличима от спорадической IPF.[3] Генетические ассоциации включают мутации в легочный сурфактант белки A1, A2, C (SFTPA1, SFTPA2B ) и муцин (MUC5B ).[22]Замечательным аспектом варианта MUC5B является его высокая частота обнаружения, так как он обнаруживается примерно у 20% людей северного и западноевропейского происхождения и у 19% популяции Framingham Heart Study.[23]Мутации у человека теломераза гены также связаны с семейным фиброзом легких и у некоторых пациентов со спорадической ИЛФ (например, TERT, TERC гены).[22] Недавно X-сцепленная мутация в третьем гене, связанном с теломеразой, дискерине (DKC1), была описана в семье с IPF.[24][ненадежный медицинский источник? ]
Диагностика
Ранняя диагностика IPF является предпосылкой для более раннего лечения и, возможно, улучшения долгосрочного клинического результата этого прогрессирующего и в конечном итоге смертельного заболевания.[3] Если есть подозрение на IPF, диагностика может быть сложной, но было показано, что мультидисциплинарный подход с участием пульмонолога, радиолога и патолога, специализирующегося на интерстициальных заболеваниях легких, повышает точность диагностики IPF.[3][25][26]
Междисциплинарное консенсусное заявление по идиопатической интерстициальной пневмонии, опубликованное Американское торакальное общество (ATS) и Европейское респираторное общество (ERS) в 2000 г. предложили конкретные основные и второстепенные критерии для установления диагноза IPF.[3] Однако в 2011 году новые упрощенные и обновленные критерии диагностики и лечения ИЛФ были опубликованы ATS, ERS совместно с Японским респираторным обществом (JRS) и Латиноамериканской торакальной ассоциацией (ALAT).[3] В настоящее время для диагностики ИЛФ необходимо:
- Исключение известных причин ILD, например, воздействия окружающей среды в быту и на рабочем месте, нарушения соединительной ткани или воздействие / токсичность лекарств.
- Наличие типичной рентгенологической картины обычная интерстициальная пневмония (UIP) на компьютерная томография высокого разрешения (КТВР).
В правильных клинических условиях можно диагностировать ИЛФ только с помощью КТВР, что устраняет необходимость в хирургической биопсии легкого.[3][7]
Дифференциальная диагностика
Признание IPF в клинической практике может быть сложной задачей, поскольку симптомы часто похожи на симптомы более распространенных заболеваний, таких как астма, хроническая обструктивная болезнь легких (ХОБЛ) и хроническая сердечная недостаточность (www.diagnoseipf.com ). Ключевой вопрос, с которым сталкиваются клиницисты: симптомы (или знаки), радиология, и исследование функции легких в совокупности соответствуют диагнозу IPF или связаны ли результаты с другим процессом. Давно признано, что пациенты с ILD, связанные с асбест контакт, наркотики (Такие как химиотерапевтический агенты или нитрофурантоин ), ревматоидный артрит и склеродермия /системный склероз может быть трудно отличить от IPF. Другие вопросы дифференциальной диагностики включают интерстициальное заболевание легких, связанное с смешанное заболевание соединительной ткани, передовой саркоидоз, хронический гиперчувствительный пневмонит, легочный Гистиоцитоз клеток Лангергана и лучевое поражение легких.[3][7]
Классификация

Идиопатический фиброз легких (ИЛФ) принадлежит к большой группе из более чем 200 заболеваний легких, известных как интерстициальные заболевания легких (ILD), которые характеризуются поражением легких интерстиций,[7] ткань между воздушными мешочками легкого. IPF - это одна из конкретных презентаций идиопатическая интерстициальная пневмония (IIP), который, в свою очередь, является разновидностью ILD, также известной как диффузное паренхиматозное заболевание легких (DPLD).[нужна цитата ]
2002 год Американское торакальное общество /Европейское респираторное общество (ATS / ERS) классификация IIP была обновлена в 2013 г.[7] В этой новой классификации есть три основные категории идиопатический интерстициальные пневмонии (МИП): основные МИП, редкие МИП и неклассифицируемые МИП. Основные МИП сгруппированы в хронический фиброзирующие IP-адреса (включая IPF и неспецифическая интерстициальная пневмония [NSIP]); ИП, связанные с курением (например, респираторный бронхиолит – интерстициальное заболевание легких [RB-ILD] и десквамативная интерстициальная пневмония [ОКУНАТЬ]); и острые / подострые ИП (т. е. криптогенная организующая пневмония [COP] и острая интерстициальная пневмония [AIP]).[7]
Для диагностики IIP необходимо исключить известные причины ILD. Примеры ILD известной причины включают: гиперчувствительный пневмонит, легочный Гистиоцитоз клеток Лангергана, асбестоз, и коллагеновая сосудистая болезнь. Однако эти нарушения часто поражают не только интерстиций, но также воздушные пространства, периферические дыхательные пути и кровеносные сосуды.[7]
Радиология
Рентген грудной клетки полезны для последующего наблюдения за пациентами с ИЛФ. К сожалению, простой рентген грудной клетки не является диагностическим, но может выявить снижение объем легких, как правило, с заметными сетчатыми интерстициальными отметинами около оснований легких.[3]

Радиологическое обследование с помощью КТВР является важным этапом в диагностике IPF. КТВР выполняется с использованием обычного компьютерный аксиальный томографический сканер без введения контрастных веществ. Оценочные срезы очень тонкие, 1-2 мм.
Типичная КТГ грудной клетки IPF демонстрирует фиброзные изменения в обоих легких с предпочтением в основании и по периферии. Согласно совместным рекомендациям ATS / ERS / JRS / ALAT 2011, HRCT является важным компонентом диагностического пути при IPF, который может идентифицировать UIP по наличию:[3]
- Ретикулярное помутнение, часто связанное с тракционные бронхоэктазы
- Соты проявляется в виде скоплений кистозных воздушных пространств, как правило, сопоставимых диаметров (3–10 мм), но иногда больших. Обычно субплевральный, характеризуется четко очерченными стенками и расположен как минимум в две линии. Обычно одной линии кист недостаточно, чтобы определить соты.
- Непрозрачность матового стекла распространены, но менее обширны, чем сетчатая
- Распределение характерно базальное и периферическое, но часто неоднородное.

Гистология
Согласно обновленным рекомендациям 2011 г., при отсутствии типичного паттерна UIP на КТВР, для уверенного диагноза требуется хирургическая биопсия легкого.[3]
Гистологические образцы для диагностики IPF должны быть взяты как минимум в трех разных местах и должны быть достаточно большими, чтобы патолог мог прокомментировать основную архитектуру легких. Небольшие биопсии, например, полученные через трансбронхиальное легкое биопсия (выполняется во время бронхоскопии) обычно для этой цели недостаточно. Следовательно, биопсии большего размера, полученные хирургическим путем через торакотомия или же торакоскопия обычно необходимы.[3][7]
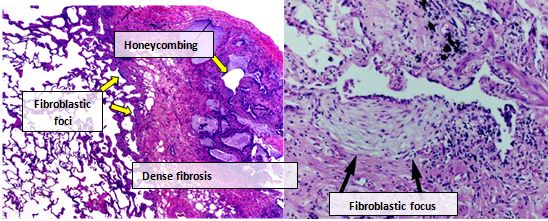
Ткань легких людей с IPF обычно показывает характерный гистопатологический паттерн UIP и, следовательно, является патологическим аналогом IPF.[3] Хотя патологический диагноз UIP часто соответствует клиническому диагнозу IPF, гистологический паттерн UIP можно увидеть и при других заболеваниях, а также при фиброзе известного происхождения (например, ревматических заболеваниях).[1][3] Есть четыре ключевых особенности UIP, включая интерстициальный фиброз в виде «лоскутного рисунка», интерстициальное рубцевание, сотовые изменения и очаги фибробластов.[нужна цитата ]
Фибробластические очаги представляют собой плотные скопления миофибробластов и рубцовой ткани и вместе с сотами являются основными патологическими находками, которые позволяют диагностировать UIP.
Бронхоальвеолярный лаваж
Бронхоальвеолярный лаваж (BAL) - это хорошо переносимая диагностическая процедура при ILD.[8] Цитологический анализ БАЛ (дифференциальный подсчет клеток) следует учитывать при оценке пациентов с ИЛФ по усмотрению лечащего врача, исходя из доступности и опыта в своем учреждении. БАЛ может выявить альтернативные специфические диагнозы: злокачественная опухоль, инфекции, эозинофильная пневмония, гистиоцитоз X, или же альвеолярный протеиноз. При оценке пациентов с подозрением на ИЛФ наиболее важным применением БАЛ является исключение других диагнозов. Видный лимфоцитоз (> 30%) в целом позволяет исключить диагноз ИЛФ.[27]
Легочные функциональные пробы
Спирометрия классически выявляет сокращение жизненная емкость (VC) либо с пропорциональным сокращением воздушных потоков, либо с увеличением воздушных потоков для наблюдаемой жизненной емкости легких. Последнее открытие отражает повышенную жесткость легких (снижение эластичности легких), связанную с фиброзом легких, что приводит к усилению упругой отдачи легких.[28]
Измерение статических объемов легких с помощью плетизмография тела или другие методы обычно выявляют уменьшение объема легких (ограничение). Это отражает трудности, возникающие при надувании фиброзных легких.
Дифференцирующая способность по окиси углерода (DLCO) неизменно снижается при IPF и может быть единственной аномалией при легкой или ранней стадии заболевания. Его нарушение лежит в основе склонности пациентов с ИЛФ к снижению насыщения кислородом при физических нагрузках, что также можно оценить с помощью теста 6-минутной ходьбы (6MWT).[3]
Такие термины, как «легкая», «умеренная» и «тяжелая» иногда используются для определения стадии заболевания и обычно основаны на измерениях функции легких в состоянии покоя.[3] Однако нет четкого консенсуса относительно стадирования пациентов с ИЛФ и того, какие критерии и значения лучше всего использовать. IPF легкой и средней степени тяжести характеризуется следующими функциональными критериями:[29][30][31][32]
- Форсированная жизненная емкость легких (ФЖЕЛ) ≥50%
- DLCO ≥30%
- Расстояние 6MWT ≥150 метров.
Уход
Цели лечения IPF заключаются в основном в уменьшении симптомов, прекращении прогрессирования заболевания, предотвращении обострений и продлении выживаемости. Профилактическую помощь (например, вакцинацию) и симптоматическое лечение следует начинать у каждого пациента на ранней стадии.[33]
Кислородная терапия
В рекомендациях IPF 2011 г. кислородная терапия, или дополнительный кислород для домашнего использования, стал настоятельной рекомендацией для пациентов со значительно низким уровнем кислорода в состоянии покоя. Хотя кислородная терапия не показала улучшения выживаемости при IPF, некоторые данные указывают на улучшение переносимости упражнений.[3][34]
Легочная реабилитация
Усталость и потеря мышечной массы являются обычными проблемами, ведущими к инвалидности у пациентов с ИЛФ. Легочная реабилитация может облегчить явные симптомы IPF и улучшить функциональный статус, стабилизируя и / или обращая вспять внелегочные признаки заболевания.[35][36] Количество опубликованных исследований роли легочной реабилитации при идиопатическом фиброзе легких невелико, но большинство из этих исследований выявили значительные краткосрочные улучшения функциональной переносимости физической нагрузки, качества жизни и одышки при физической нагрузке.[37] Типичные программы реабилитации включают физические упражнения, регулирование режима питания, трудотерапию, образование и психосоциальные консультации. В поздней фазе заболевания пациенты с ИЛФ склонны прекращать физическую активность из-за усиливающейся одышки. По возможности этого не следует поощрять.[нужна цитата ]
Лекарства
В прошлом был исследован ряд методов лечения ИЛФ, включая интерферон гамма-1β,[38] бозентан,[39] амбризентан,[40] и антикоагулянты,[41] но они больше не считаются эффективными вариантами лечения. Многие из этих ранних исследований основывались на гипотезе о воспалительном заболевании IPF.
Пирфенидон
А Кокрановский обзор сравнение пирфенидон с плацебо было обнаружено снижение риска прогрессирования заболевания на 30%.[42] FVC или VC также были улучшены, даже если небольшое замедление снижения FVC могло быть продемонстрировано только в одном из двух испытаний CAPACITY.[29] Третье исследование, которое было завершено в 2014 году, показало снижение снижения функции легких и прогрессирования заболевания IPF.[31] Данные исследования ASCEND также были объединены с данными двух исследований CAPACITY в предварительно заданном анализе, который показал, что пирфенидон снижает риск смерти почти на 50% в течение одного года лечения.[31]
N-ацетилцистеин и тройная терапия
N-Ацетилцистеин (NAC) является предшественником глутатиона, антиоксидант. Было высказано предположение, что лечение высокими дозами NAC может восстановить дисбаланс между оксидантами и антиоксидантами, который возникает в ткани легких пациентов с IPF. В первом клиническом исследовании с участием 180 пациентов (IFIGENIA) в предыдущем исследовании было показано, что NAC снижает снижение VC и DLCO в течение 12 месяцев наблюдения при использовании в сочетании с преднизон и азатиоприн (тройная терапия).[43]
Совсем недавно большое рандомизированное контролируемое исследование (PANTHER-IPF) было проведено Национальные институты здоровья (NIH) в США для оценки тройной терапии и монотерапии NAC у пациентов с IPF. Это исследование показало, что комбинация преднизона, азатиоприна и NAC увеличивает риск смерти и госпитализации.[44] а в 2012 году NIH объявил, что группа тройной терапии в исследовании PANTHER-IPF была досрочно прекращена.[45]
В этом исследовании также оценивали только NAC, и результаты этой части исследования были опубликованы в мае 2014 года в Медицинском журнале Новой Англии, в котором сделан вывод о том, что «по сравнению с плацебо ацетилцистеин не дает значительного преимущества в отношении сохранения FVC у пациентов. с идиопатическим легочным фиброзом с легкими или умеренными нарушениями функции легких ».[46]
Нинтеданиб
Нинтеданиб это тройка ингибитор ангиокиназы это нацелено рецепторные тирозинкиназы участвует в регулировании ангиогенез: рецептор фактора роста фибробластов (FGFR), рецептор фактора роста тромбоцитов (PDGFR) и рецептор фактора роста эндотелия сосудов (VEGFR),[47] которые также участвуют в патогенезе фиброза и IPF. В обоих исследованиях фазы III нинтеданиб уменьшал снижение функции легких примерно на 50% в течение одного года.[32] Он был одобрен FDA США в октябре 2014 г.[48] и авторизован в Европе в январе 2015 года.[49]
Трансплантация легких
Трансплантация легких может быть подходящим для тех пациентов, которые физически подходят для серьезной операции по трансплантации. Было показано, что у пациентов с ИЛФ трансплантация легких снижает риск смерти на 75% по сравнению с пациентами, которые остаются в списке ожидания.[50] С момента введения оценка распределения легких (LAS), который отдает приоритет кандидатам на трансплантацию на основе вероятности выживания, IPF стал наиболее распространенным показанием для трансплантации легких в США.[35]
Симптоматические пациенты с ИЛФ в возрасте до 65 лет и с индексом массы тела (ИМТ) ≤26 кг / м2 следует направить на трансплантацию легких, но нет четких данных, которые позволили бы определить точное время для LTx. Несмотря на противоречие, самые последние данные предполагают, что двусторонняя трансплантация легкого превосходит трансплантацию одного легкого у пациентов с ИЛФ.[51] Пятилетняя выживаемость после трансплантации легкого в IPF оценивается от 50 до 56%.[3][52][53]
Паллиативная помощь
Паллиативная помощь фокусируется на уменьшении симптомов и повышении комфорта пациентов, а не на лечении болезни. Это может включать лечение обострения симптомов с использованием хронических опиоиды при сильной одышке и кашле. Кроме того, кислородная терапия может быть полезной для облегчения одышки у пациентов с гипоксемией.
Паллиативная помощь также включает облегчение физических и эмоциональных страданий и психологическую поддержку пациентов и лиц, осуществляющих уход.[3] По мере прогрессирования заболевания пациенты могут испытывать страх, тревогу и депрессию, поэтому следует рассмотреть возможность психологического консультирования. В недавнем исследовании амбулаторных пациентов с ILD, включая IPF, оценку депрессии, функциональный статус (оцениваемый с помощью теста ходьбы), а также легочную функцию, все они влияли на тяжесть одышки.[54]
В отдельных случаях особенно тяжелой одышки морфий можно было бы рассмотреть. Он может уменьшить одышку, беспокойство и кашель без значительного снижения сатурации кислорода.[55]
Следовать за
IPF часто ошибочно диагностируется, по крайней мере, до тех пор, пока физиологические данные и / или данные изображений не предполагают наличие ILD, ведущего к задержке обращения за соответствующей помощью.[35] Учитывая, что IPF - это заболевание, средняя выживаемость которого составляет три года после постановки диагноза, поэтому следует рассмотреть возможность раннего направления в специализированный центр для любого пациента с подозрением или известным ILD. На основе комплексной дифференциальной диагностики, многопрофильное обсуждение между пульмонологами, радиологами и патологами, имеющими опыт диагностики ILD, имеет первостепенное значение для точного диагноза.[3]
После постановки диагноза IPF и выбора подходящего лечения в соответствии с симптомами и стадией заболевания необходимо тщательное наблюдение. Из-за высокой вариабельности течения заболевания, более высокой частоты таких осложнений, как рак легких (до 25% пациентов были зарегистрированы в IPF), плановая оценка каждые 3-6 месяцев, включая спирометрию (плетизмографию тела), тестирование диффузионной способности , рентген грудной клетки, 6MWT, оценка одышки, качества жизни, потребность в кислороде обязательна.[нужна цитата ]
Кроме того, растущая осведомленность об осложнениях и общих сопутствующих состояниях, часто связанных с ИЛФ, требует регулярной оценки сопутствующих заболеваний, большинство из которых просто отражает сопутствующие заболевания старения, а также лекарства с их взаимодействием и побочными эффектами.
Острые обострения
Острые обострения IPF (AE-IPF) определяются как необъяснимое ухудшение или развитие одышки в течение 30 дней с новыми рентгенологическими инфильтратами при аномалии HRCT, часто наложенной на фон, соответствующий паттерну UIP. Ежегодная частота AE-IPF составляет от 10 до 15% всех пациентов. Прогноз AE-IPF плохой, летальность составляет от 78% до 96%.[56] Необходимо исключить другие причины AE-IPF, такие как тромбоэмболия легочной артерии, застойная сердечная недостаточность, пневмоторакс или инфекция. Легочную инфекцию следует исключить с помощью эндотрахеального аспирата или БАЛ.
Многие пациенты с острым ухудшением состояния нуждаются в интенсивной терапии, особенно когда дыхательная недостаточность связана с гемодинамической нестабильностью, значительными сопутствующими заболеваниями или тяжелой гипоксемией.[57] Однако летальность при госпитализации высока.[56] Механическую вентиляцию легких следует вводить только после тщательного анализа долгосрочного прогноза пациента и, по возможности, его желаний. Однако современные руководства не рекомендуют использовать механическую вентиляцию легких у пациентов с дыхательной недостаточностью, вызванной ИЛФ.[3]
Прогноз
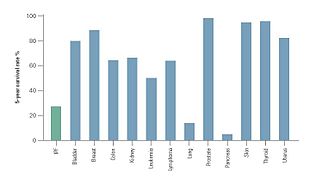
Клиническое течение IPF может быть непредсказуемым.[3][58][59] Прогрессирование IPF связано с оценкой среднего времени выживания от 2 до 5 лет после постановки диагноза.[1][3]5-летняя выживаемость для IPF составляет 20-40%,[59] уровень смертности выше, чем от ряда злокачественных новообразований, включая рак толстой кишки, множественную миелому и рак мочевого пузыря.[58][59]
Недавно для прогнозирования смертности при ИЛФ был предложен многомерный индекс и система стадий.[60] Название индекса - GAP, он основан на поле [G], возрасте [A] и двух переменных физиологии легких [P] (FVC и DL.CO которые обычно измеряются в клинической практике для прогнозирования смертности при IPF. Установлено, что высшая стадия ГАП (стадия III) связана с 39% риском смерти через 1 год.[60] Эта модель также была оценена в IPF и других ILD и показала хорошие результаты в прогнозировании смертности во всех основных подтипах ILD. Модифицированный индекс ILD-GAP был разработан для применения к подтипам ILD с целью получения оценок выживаемости при конкретных заболеваниях.[61] У пациентов с ИЛФ общая смертность через 5 лет высока, но годовой уровень смертности от всех причин у пациентов с легкими поражениями легкой и средней степени тяжести относительно низок. Это причина, по которой изменение функции легких (ФЖЕЛ) обычно измеряется в ходе однолетних клинических испытаний лечения ИЛФ, а не выживаемости.[62]
В дополнение к клиническим и физиологическим параметрам, позволяющим предсказать, насколько быстро могут прогрессировать пациенты с IPF, генетические и молекулярные особенности также связаны со смертностью от IPF. Например, было показано, что пациенты с IPF, которые имеют конкретный генотип в полиморфизме гена муцина MUC5B (см. Выше), испытывают более медленное снижение FVC и значительно улучшают выживаемость.[63][64] Даже если такие данные интересны с научной точки зрения, применение в клинической практике прогностической модели, основанной на конкретных генотипах, по-прежнему невозможно.
Эпидемиология
IPF, хотя и встречается редко, является наиболее распространенной формой IIP.[7] Распространенность ИЛФ оценивается от 14,0 до 42,7 на 100 000 человек на основе анализа данных о заявках на медицинское обслуживание в США, с вариациями в зависимости от определений случая, использованных в этом анализе.[9][65] IPF чаще встречается у мужчин, чем у женщин, и обычно диагностируется у людей старше 50 лет.[3]
В заболеваемость IPF трудно определить, поскольку единообразные диагностические критерии не применялись последовательно.[3][9] Согласно недавнему исследованию, проведенному в США, частота ИЛФ составляет от 6,8 до 16,3 на 100 000 человек. В 27 странах Европейского Союза, по данным ряда источников, заболеваемость составляет 4,6–7,4 человек на 100 000 населения,[66][67] это означает, что ежегодно около 30 000–35 000 новых пациентов будут диагностироваться с ИЛФ.[65][68]
Недавнее одноцентровое ретроспективное обсервационное когортное исследование, в котором участвовали пациенты с диагнозом ИЛЗ в Больница Орхусского университета (Дания) в период с 2003 по 2009 год выявили заболеваемость ИЛЗ 4,1 на 100 000 жителей в год. IPF был наиболее частым диагнозом (28%), за ним следовали ILD, связанные с заболеванием соединительной ткани (14%), гиперчувствительный пневмонит (7%) и неспецифическая интерстициальная пневмония (NSIP) (7%). Заболеваемость ИЛФ составила 1,3 на 100 000 жителей в год.[69]
В связи с неоднородным распределением болезни по европейским странам эпидемиологические данные необходимо обновлять через общеевропейский регистр ILD и IPF.
Другие животные
IPF был признан у нескольких пород собак и кошек,[70] и был лучше всего охарактеризован в Вест-хайленд-уайт-терьеры.[71] Ветеринарные пациенты с этим заболеванием имеют многие из тех же клинических признаков, что и их коллеги-люди, включая прогрессирующую непереносимость физических упражнений, учащенное дыхание и возможный респираторный дистресс.[72]Прогноз в целом плохой.
Исследование
Ряд агентов в настоящее время расследуются в Фаза II клинических испытаний для IPF, включая моноклональные антитела симтузумаб, тралокинумаб, лебрикизумаб и FG-3019, а лизофосфатидная кислота антагонист рецепторов (BMS-986020). Также продолжается исследование фазы II STX-100.[73] Эти молекулы направлены против нескольких факторов роста и цитокинов, которые, как известно, играют роль в пролиферации, активации, дифференцировке или ненадлежащем выживании фибробластов.[нужна цитата ]
предшественник микроРНК mir-29 Исследования на мышах показали обратимость индуцированной IPF. MRG-201 в настоящее время проходит испытания по состоянию на 2016 год, но еще не на пациентах с IPF, и на январь 2016 года испытания на людях для использования IPF не запланированы.[Обновить].[74]
Лечение стволовыми клетками для IPF - это область исследований.[75][76]
Рекомендации
- ^ а б c d е ж грамм час я j k л м п о п q р «Идиопатический фиброз легких». NHLBI. Получено 21 января 2018.
- ^ а б c d Рагху Г., Рохверг Б., Чжан И, Гарсия, Калифорния, Адзума А., Бер Дж. И др. (Июль 2015 г.). «Официальное руководство по клинической практике ATS / ERS / JRS / ALAT: лечение идиопатического фиброза легких. Обновление Руководства по клинической практике 2011 года». Американский журнал респираторной медицины и реанимации. 192 (2): e3–19. Дои:10.1164 / rccm.201506-1063ST. PMID 26177183.
- ^ а б c d е ж грамм час я j k л м п о п q р s т ты v ш Икс у z аа ab ac объявление ае аф аг ах ай Рагху Дж., Коллард Х. Р., Иган Дж. Дж. И др. (2011). «Официальное заявление ATS / ERS / JRS / ALAT: Идиопатический фиброз легких: научно обоснованные рекомендации по диагностике и лечению». Американский журнал респираторной медицины и реанимации. 183 (6): 788–824. Дои:10.1164 / rccm.2009-040GL. ЧВК 5450933. PMID 21471066.
- ^ а б c d е ж Ферри, Фред Ф. (2017). Электронная книга Ferri's Clinical Advisor 2018: 5 книг в 1. Elsevier Health Sciences. п. 691. ISBN 9780323529570.
- ^ «Идиопатический фиброз легких | NHLBI, NIH». www.nhlbi.nih.gov. Получено 2020-12-05.
- ^ а б Мельцер Э.Б., Благородный П.В. (2008). «Идиопатический фиброз легких». Журнал редких заболеваний Orphanet. 3 (1): 8. Дои:10.1186/1750-1172-3-8. ЧВК 2330030. PMID 18366757.
- ^ а б c d е ж грамм час я j Трэвис У.Д., Костабель У., Ханселл Д.М., Кинг Т.Е., Линч Д.А., Николсон А.Г. и др. (Сентябрь 2013). «Официальное заявление Американского торакального общества / Европейского респираторного общества: обновление международной мультидисциплинарной классификации идиопатических интерстициальных пневмоний». Американский журнал респираторной медицины и реанимации. 188 (6): 733–48. Дои:10.1164 / rccm.201308-1483ST. ЧВК 5803655. PMID 24032382.
- ^ а б Флаэрти К.Р., Мамфорд Дж. А., Мюррей С., Казеруни Е. А., Гросс Б. Х., Колби ТВ, Трэвис В. Д., Флинт А. и др. (2007). «Прогностические последствия физиологических и рентгенологических изменений при идиопатической интерстициальной пневмонии». Американский журнал респираторной медицины и реанимации. 168 (5): 543–548. CiteSeerX 10.1.1.320.6411. Дои:10.1164 / rccm.200209-1112OC. PMID 12773329.
- ^ а б c Рагху Г., Вейкер Д., Эдесберг Дж., Брэдфорд В.З., Остер Дж. (2006). «Заболеваемость и распространенность идиопатического фиброза легких». Американский журнал респираторной медицины и реанимации. 174 (7): 810–816. Дои:10.1164 / rccm.200602-163oc. PMID 16809633.
- ^ а б Коттин V, Кордье Дж. Ф. (2012). «Хрипы на липучках: ключ к ранней диагностике идиопатического фиброза легких». Европейский респираторный журнал. 40 (3): 519–521. Дои:10.1183/09031936.00001612. PMID 22941541.
- ^ Баумэн Р.П., Шипли Р.Т., Лаудон Р.Г., Нижний Восточный Восток (1991). «Хрипы при интерстициальной болезни легких. Сравнение саркоидоза и фиброзирующего альвеолита». Грудь. 100 (1): 96–101. Дои:10.1378 / сундук.100.1.96. PMID 2060395.
- ^ а б c Олсон А. Л., Свигрис Дж. Дж. (Март 2012 г.). «Идиопатический фиброз легких: диагностика и эпидемиология». Клиники грудной медицины. 33 (1): 41–50. Дои:10.1016 / j.ccm.2011.12.001. PMID 22365244.
- ^ Уильямс, KJ (март 2014 г.). «Гаммахерпесвирусы и фиброз легких: данные от людей, лошадей и грызунов». Ветеринарная патология. 51 (2): 372–384. Дои:10.1177/0300985814521838. PMID 24569614. S2CID 22704874.
- ^ Гарсия-Санчо С., Буэндиа-Рольдан I, Фернандес-Плата М.Р., Наварро С., Перес-Падилья Р., Варгас М.Х. и др. (Декабрь 2011 г.). «Семейный фиброз легких - самый сильный фактор риска идиопатического фиброза легких». Респираторная медицина. 105 (12): 1902–7. Дои:10.1016 / j.rmed.2011.08.022. PMID 21917441.[ненадежный медицинский источник? ]
- ^ а б Харари С., Каминати А. (2010). «IPF: новый взгляд на патогенез и лечение». Аллергия. 65 (5): 537–553. Дои:10.1111 / j.1398-9995.2009.02305.x. PMID 20121758.
- ^ Джастис Дж. Н., Намбияр А. М., Чкония Т., Киркланд Дж. Л. (2019). «Сенолитики при идиопатическом фиброзе легких: результаты первого открытого пилотного исследования с участием людей». EBioMedicine. 40: 554–563. Дои:10.1016 / j.ebiom.2018.12.052. ЧВК 6412088. PMID 30616998.
- ^ Палмер А.К., Густафсон Б., Киркланд Д.Л., Смит Ю. (2019). «Клеточное старение: связь между старением и диабетом». Диабетология. 62 (10): 1835–1841. Дои:10.1007 / s00125-019-4934-х. ЧВК 6731336. PMID 31451866.
- ^ Киркланд JL, Tchkonia T (2020). «Сенолитические препараты: от открытия до перевода». Журнал внутренней медицины. Дои:10.1111 / joim.13141. ЧВК 7405395. PMID 32686219.
- ^ а б Лумис-Кинг Х, Флаэрти К. Р., Мур Б. Б. (апрель 2013 г.). «Патогенез, современные методы лечения и будущие направления лечения идиопатического фиброза легких». Текущее мнение в фармакологии. 13 (3): 377–385. Дои:10.1016 / j.coph.2013.03.015. ЧВК 3686907. PMID 23602652.
- ^ Пардо А, Селман М (2002). «Идиопатический фиброз легких: новые взгляды на его патогенез». Международный журнал биохимии и клеточной биологии. 34 (12): 1534–1538. Дои:10.1016 / с 1357-2725 (02) 00091-2. PMID 12379275.
- ^ Селман М., Кинг Т.Э., Пардо А (2001). «Идиопатический фиброз легких: преобладающие и развивающиеся гипотезы о его патогенезе и последствиях для терапии». Анналы внутренней медицины. 134 (2): 136–151. Дои:10.7326/0003-4819-134-2-200101160-00015. PMID 11177318. S2CID 10955241.
- ^ а б «Запись OMIM - № 178500 - ФИБРОЗ ЛЕГКИ, ИДИОПАТИЧЕСКИЙ; IPF». Omim.org. Получено 7 июн 2018.
- ^ Mathai S, et al. (2014). «Генетическая предрасположенность и фиброз легких». Текущее мнение в области легочной медицины. 20 (5): 429–435. Дои:10.1097 / MCP.0000000000000074. ЧВК 4337021. PMID 25022318.
- ^ Кропски Дж. А., Митчелл Д. Б., Маркин С. и др. (6 февраля 2014 г.). «Новая мутация дискерина (DKC1) связана с семейной интерстициальной пневмонией». Грудь. 146 (1): e1–7. Дои:10.1378 / сундук.13-2224. ЧВК 4077414. PMID 24504062.
- ^ Флаэрти KR, King TE, Raghu G, Lynch JP, Colby TV, Travis WD, Gross BH, Kazerooni EA и др. (2004). «Идиопатическая интерстициальная пневмония: каков эффект мультидисциплинарного подхода к диагностике?». Американский журнал респираторной медицины и реанимации. 170 (8): 904–910. Дои:10.1164 / rccm.200402-147OC. PMID 15256390.
- ^ Флаэрти KR, Андрей AC, King TE Jr, Raghu G, Colby TV, Wells A, Bassily N, Brown K и др. (2007). «Идиопатическая интерстициальная пневмония: согласны ли общественные и академические врачи по поводу диагноза?». Американский журнал респираторной медицины и реанимации. 175 (10): 1054–1060. Дои:10.1164 / rccm.200606-833OC. ЧВК 1899268. PMID 17255566.
- ^ Охшимо С., Бонелла Ф., Цуй А., Бьюме М., Коно Н., Гусман Дж., Костабель Ю. (2009). «Значение бронхоальвеолярного лаважа для диагностики идиопатического фиброза легких». Американский журнал респираторной медицины и реанимации. 179 (11): 1043–1047. Дои:10.1164 / rccm.200808-1313oc. PMID 19246718.
- ^ Pellegrino R, Viegi G, Brusasco V, Crapo RO, Burgos F, Casaburi R, Coates A, van der Grinten CP, et al. (2005). "Interpretative strategies for lung function tests". European Respiratory Journal. 26 (5): 948–968. Дои:10.1183/09031936.05.00035205. PMID 16264058.
- ^ а б Noble PW, Albera C, Bradford WZ, Costabel U, Glassberg MK, Kardatzke D, King TE, Lancaster L, et al. (2011). "Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials". Ланцет. 377 (9779): 1760–1769. Дои:10.1016/S0140-6736(11)60405-4. PMID 21571362. S2CID 10119356.
- ^ Martinez FJ, Safrin S, Weycker D, Starko KM, Bradford WZ, King TE, Flaherty KR, Schwartz DA, Noble PW, Raghu G, Brown KK (June 2005). "The clinical course of patients with idiopathic pulmonary fibrosis". Анналы внутренней медицины. 142 (12 Pt 1): 963–7. Дои:10.7326/0003-4819-142-12_part_1-200506210-00005. PMID 15968010. S2CID 24224976.
- ^ а б c King TE, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, et al. (Май 2014 г.). "A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis" (PDF). Медицинский журнал Новой Англии. 370 (22): 2083–92. Дои:10.1056/NEJMoa1402582. PMID 24836312.
- ^ а б Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. (Май 2014 г.). "Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis" (PDF). Медицинский журнал Новой Англии. 370 (22): 2071–82. Дои:10.1056/NEJMoa1402584. HDL:11365/974374. PMID 24836310.
- ^ Lee JS, McLaughlin S, Collard HR (2011). "Comprehensive care of the patient with idiopathic pulmonary fibrosis". Текущее мнение в области легочной медицины. 17 (5): 348–354. Дои:10.1097/mcp.0b013e328349721b. PMID 21760508. S2CID 11918582.
- ^ Morrison DA, Stovall JR (1992). "Increased exercise capacity in hypoxemic patients after long-term oxygen therapy". Грудь. 102 (2): 542–550. Дои:10.1378/chest.102.2.542. PMID 1643945.
- ^ а б c Spagnolo P, Tonelli R, Cocconcelli E, Stefani A, Richeldi L (2012). "Idiopathic pulmonary fibrosis: diagnostic pitfalls and therapeutic challenges". Multidisciplinary Respiratory Medicine. 7 (1): 42. Дои:10.1186/2049-6958-7-42. ЧВК 3537555. PMID 23146172.
- ^ Lee JS, McLaughlin S, Collard HR (September 2011). "Comprehensive care of the patient with idiopathic pulmonary fibrosis". Текущее мнение в области легочной медицины. 17 (5): 348–54. Дои:10.1097/mcp.0b013e328349721b. PMID 21760508. S2CID 11918582.
- ^ Kenn K, Gloeckl R, Behr J (2013). "Pulmonary rehabilitation in patients with idiopathic pulmonary fibrosis--a review". Дыхание; Международный обзор заболеваний грудной клетки. 86 (2): 89–99. Дои:10.1159/000354112. PMID 23942353.
- ^ King TE, Albera C, Bradford WZ, Costabel U, Hormel P, Lancaster L, et al. (Июль 2009 г.). "Effect of interferon gamma-1b on survival in patients with idiopathic pulmonary fibrosis (INSPIRE): a multicentre, randomised, placebo-controlled trial". Ланцет. 374 (9685): 222–8. Дои:10.1016/S0140-6736(09)60551-1. PMID 19570573. S2CID 2432490.
- ^ King TE, Brown KK, Raghu G, du Bois RM, Lynch DA, Martinez F, et al. (Июль 2011 г.). "BUILD-3: a randomized, controlled trial of bosentan in idiopathic pulmonary fibrosis". Американский журнал респираторной медицины и реанимации. 184 (1): 92–9. Дои:10.1164/rccm.201011-1874OC. PMID 21474646.
- ^ Raghu G, Behr J, Brown KK, Egan JJ, Kawut SM, Flaherty KR, et al. (Май 2013). "Treatment of idiopathic pulmonary fibrosis with ambrisentan: a parallel, randomized trial". Анналы внутренней медицины. 158 (9): 641–9. Дои:10.7326/0003-4819-158-9-201305070-00003. PMID 23648946.
- ^ Noth I, Anstrom KJ, Calvert SB, de Andrade J, Flaherty KR, Glazer C, et al. (Июль 2012 г.). "A placebo-controlled randomized trial of warfarin in idiopathic pulmonary fibrosis". Американский журнал респираторной медицины и реанимации. 186 (1): 88–95. Дои:10.1164/rccm.201202-0314OC. ЧВК 3400994. PMID 22561965.
- ^ Spagnolo P, Del Giovane C, Luppi F, Cerri S, Balduzzi S, Walters EH, D'Amico R, Richeldi L (2010). "Non-steroid agents for idiopathic pulmonary fibrosis". Кокрановская база данных систематических обзоров (9): CD003134. Дои:10.1002/14651858.CD003134.pub2. HDL:11380/680648. PMID 20824834.
- ^ Demedts M, Behr J, Buhl R, Costabel U, Dekhuijzen R, Jansen HM, MacNee W, Thomeer M, et al. (2005). "High-dose acetylcysteine in idiopathic pulmonary fibrosis" (PDF). Медицинский журнал Новой Англии. 353 (21): 2229–2242. Дои:10.1056/NEJMoa042976. HDL:2066/47718. PMID 16306520.
- ^ Raghu G, Anstrom KJ, King TE, Lasky JA, Martinez FJ (May 2012). "Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis". Медицинский журнал Новой Англии. 366 (21): 1968–77. Дои:10.1056/NEJMoa1113354. ЧВК 3422642. PMID 22607134.
- ^ "Commonly used three-drug regimen for idiopathic pulmonary fibrosis found harmful". Национальные институты здравоохранения США. 21 октября 2011 г.. Получено 2013-04-11.
- ^ The Idiopathic Pulmonary Fibrosis Clinical Research Network. (2014). "Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis". Медицинский журнал Новой Англии. 370 (22): 2093–2102. Дои:10.1056/nejmoa1401739. ЧВК 4116664. PMID 24836309.
- ^ "BIBF 1120 Fact Sheet" (PDF). Dl.groovygecko.net. Получено 2014-04-08.
- ^ "FDA Approval Package for Nintedanib" (PDF). www.accessdata.fda.gov. Получено 2019-01-07.
- ^ "Ofev | European Medicines Agency". www.ema.europa.eu. 2018-09-17. Получено 2019-01-07.
- ^ Russo MJ, Iribarne A, Hong KN, Davies RR, Xydas S, Takayama H, Ibrahimiye A, Gelijns AC, Bacchetta MD, D'Ovidio F, Arcasoy S, Sonett JR (2010). "High lung allocation score is associated with increased morbidity and mortality following transplantation". Грудь. 137 (3): 651–657. Дои:10.1378/chest.09-0319. ЧВК 2832864. PMID 19820072.
- ^ George TJ, Arnaoutakis GJ, Shah AS (2007). "Lung transplantation for idiopathic pulmonary fibrosis". Летопись торакальной хирургии. 84 (4): 1121–1128. Дои:10.1016/j.athoracsur.2007.04.096. PMID 17888957.
- ^ Mason DP, Brizzio ME, Alster JM, McNeill AM, Murthy SC, Budev MM, Mehta AC, Minai OA, et al. (2011). "Lung transplant in idiopathic pulmonary fibrosis". Архив хирургии. 146 (10): 1204–1209. Дои:10.1001/archsurg.2011.239. PMID 22006881.
- ^ Keating D, Levvey B, Kotsimbos T, Whitford H, Westall G, Williams T, Snell G (2009). "Lung transplantation in pulmonary fibrosis challenging early outcomes counter balanced by surprisingly good outcomes beyond 15 years". Трансплантация. 41 (1): 289–291. Дои:10.1016/j.transproceed.2008.10.042. PMID 19249537.
- ^ Ryerson CJ, Berkeley J, Carrieri-Kohlman VL, Pantilat SZ, Landefeld CS, Collard HR (2011). "Depression and functional status are strongly associated with dyspnea in interstitial lung disease" (PDF). Грудь. 139 (3): 609–616. Дои:10.1378/chest.10-0608. PMID 20688924. S2CID 34116718.
- ^ Allen S, Raut S, Woollard J, Vassallo M (2005). "Low dose diamorphine reduces breathlessness without causing a fall in oxygen saturation in elderly patients with end-stage idiopathic pulmonary fibrosis". Palliative Medicine. 19 (2): 128–130. Дои:10.1191/0269216305pm998oa. PMID 15810751. S2CID 12999693.
- ^ а б Agarwal R, Jindal SK (2008). "Acute exacerbation of idiopathic pulmonary fibrosis: a systematic review". Европейский журнал внутренней медицины. 19 (4): 227–235. Дои:10.1016/j.ejim.2007.04.024. PMID 18471669.
- ^ Stern JB, Mal H, Groussard O, Brugière O, Marceau A, Jebrak G, Fournier M (2001). "Prognosis of patients with advanced idiopathic pulmonary fibrosis requiring mechanical ventilation for acute respiratory failure". Грудь. 120 (1): 213–219. Дои:10.1378/chest.120.1.213. PMID 11451841.
- ^ а б c Bjoraker JA, Ryu JH, Edwin MK, Myers JL, Tazelaar HD, Schroeder DR, Offord KP (1998). "Prognostic significance of histopathologic subsets in idiopathic pulmonary fibrosis" (PDF). Американский журнал респираторной медицины и реанимации. 157 (1): 199–203. Дои:10.1164/ajrccm.157.1.9704130. PMID 9445300. S2CID 13942321.
- ^ а б c Kim DS, Collard HR, King TE (June 2006). "Classification and natural history of the idiopathic interstitial pneumonias". Труды Американского торакального общества. 3 (4): 285–92. Дои:10.1513/pats.200601-005TK. ЧВК 2658683. PMID 16738191.
- ^ а б Ley B, Ryerson CJ, Vittinghoff E, Ryu JH, Tomassetti S, Lee JS, Poletti V, Buccioli M, Elicker BM, Jones KD, King TE Jr, Collard HR (2012). "A multidimensional index and staging system for idiopathic pulmonary fibrosis". Анналы внутренней медицины. 156 (10): 684–691. CiteSeerX 10.1.1.691.4472. Дои:10.7326/0003-4819-156-10-201205150-00004. PMID 22586007. S2CID 207536377.
- ^ Ryerson CJ, Vittinghoff E, Ley B, Lee JS, Mooney JJ, Jones KD, Elicker BM, Wolters PJ, et al. (2014). "Predicting Survival Across Chronic Interstitial Lung Disease: The ILD-GAP Model". Грудь. 145 (4): 723–728. Дои:10.1378/chest.13-1474. PMID 24114524.
- ^ King TE, Albera C, Bradford WZ, Costabel U, du Bois RM, Leff JA, Nathan SD, Sahn SA, Valeyre D, Noble PW (April 2014). "All-cause mortality rate in patients with idiopathic pulmonary fibrosis. Implications for the design and execution of clinical trials". Американский журнал респираторной медицины и реанимации. 189 (7): 825–31. Дои:10.1164/rccm.201311-1951OC. HDL:2318/156709. PMID 24476390.
- ^ Peljto AL, Zhang Y, Fingerlin TE, Ma SF, Garcia JG, Richards TJ, Silveira LJ, Lindell KO, et al. (2013). "Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis". JAMA. 309 (21): 2232–2239. Дои:10.1001/jama.2013.5827. ЧВК 4545271. PMID 23695349.
- ^ Stock CJ, Sato H, Fonseca C, Banya WA, Molyneaux PL, Adamali H, Russell AM, Denton CP, et al. (2013). "Mucin 5B promoter polymorphism is associated with idiopathic pulmonary fibrosis but not with development of lung fibrosis in systemic sclerosis or sarcoidosis". Грудная клетка. 68 (5): 436–441. Дои:10.1136/thoraxjnl-2012-201786. PMID 23321605.
- ^ а б Pulmonary Fibrosis Foundation. "Prevalence and Incidence". Pulmonaryfibrosis.org. Retrieved 2013-04-11
- ^ Gribbin J, Hubbard RB, Le Jeune I, Smith CJ, West J, Tata LJ (2006). "Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK". Грудная клетка. 61 (11): 980–985. Дои:10.1136/thx.2006.062836. ЧВК 2121155. PMID 16844727.
- ^ "Eurostat News Release. European demography. 110/2010. 27 July 2010" (PDF). Epp.eurostat.ec.europa.eu. Получено 7 июн 2018.
- ^ Hyldgaard C, Hilberg O, Muller A, Bendstrup E (2014). "A cohort study of interstitial lung diseases in central Denmark". Респираторная медицина. 108 (5): 793–799. Дои:10.1016/j.rmed.2013.09.002. PMID 24636811.
- ^ Williams K, Malarkey D, Cohn L, Patrick D, Dye J, Toews G (2004). "Identification of spontaneous feline idiopathic pulmonary fibrosis: morphology and ultrastructural evidence for a type II pneumocyte defect". Грудь. 125 (6): 2278–2288. Дои:10.1378/chest.125.6.2278. PMID 15189952.
- ^ Webb JA, Armstrong J (2002). "Chronic idiopathic pulmonary fibrosis in a West Highland white terrier". Канадский ветеринарный журнал. 43 (9): 703–705. ЧВК 339552. PMID 12240528.
- ^ "AKC Canine Health Foundation". Akcchf.org. Получено 7 июн 2018.
- ^ "Active Clinical Trials and Investigational Research in IPF". Архивировано из оригинал на 2014-09-04. Получено 2014-09-04.
- ^ "Research Demonstrates Reversal Of Pulmonary Fibrosis With miRagen Therapeutics Synthetic microRNA-29 Mimic (promiR-29)". Pulmonaryfibrosisnews.com. 2014-09-23. Получено 8 июн 2018.
- ^ Liu M, Ren D, Wu D, Zheng J, Tu W (2015). "Stem Cell and Idiopathic Pulmonary Fibrosis: Mechanisms and Treatment". Текущие исследования стволовых клеток и терапия. 10 (6): 466–76. Дои:10.2174/1574888X10666150519092639. PMID 25986617.
- ^ "Stem cell therapy for lung fibrosis conditions". Sciencedaily.com. Получено 8 июн 2018.
внешняя ссылка
| Классификация | |
|---|---|
| Внешние ресурсы |