Спинальная мышечная атрофия - Spinal muscular atrophy
| Спинальная мышечная атрофия | |
|---|---|
| Другие имена | Аутосомно-рецессивная проксимальная мышечная атрофия позвоночника, спинальная мышечная атрофия 5q |
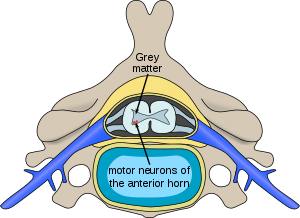 | |
| Расположение нейронов, пораженных спинальной мышечной атрофией, в спинном мозге | |
| Специальность | Неврология |
| Симптомы | Прогрессирующая мышечная слабость[1] |
| Осложнения | Сколиоз, совместные контрактуры, пневмония[2] |
| Типы | Тип 0 - тип 4[2] |
| Причины | Мутация в SMN1[2] |
| Диагностический метод | Генетическое тестирование[1] |
| Дифференциальная диагностика | Врожденная мышечная дистрофия, Мышечная дистрофия Дюшенна, Синдром Прадера-Вилли[2] |
| Уход | Поддерживающая терапия, лекарства[1] |
| Медикамент | Нусинерсен, онасемноген абепарвовец, Рисдиплам |
| Прогноз | Зависит от типа[2] |
| Частота | 1 из 10000 человек[2] |
Спинальная мышечная атрофия (SMA) является редким нервно-мышечное расстройство что приводит к потере двигательные нейроны и прогрессивный атрофия мышц.[3][4][5] Его обычно диагностируют в младенчестве или раннем детстве, и если его не лечить, это наиболее частая генетическая причина детской смерти.[6] Он также может появиться в более позднем возрасте, а затем иметь более легкое течение болезни. Общей чертой является прогрессирующая слабость произвольных мышц рук, ног и дыхательные мышцы пострадает первым.[7][8] Сопутствующие проблемы могут включать плохой контроль головы, трудности с глотанием, сколиоз, и совместные контрактуры.[9][8]
Возраст начала и тяжесть симптомов лежат в основе традиционной классификации спинальной мышечной атрофии на несколько типов.[4]
Спинальная мышечная атрофия возникает из-за аномалии (мутация ) в SMN1 ген[10][9] который кодирует SMN, а белок необходимо для выживания двигательные нейроны.[8] Потеря этих нейронов в спинном мозге препятствует передаче сигналов между мозг и скелетные мышцы.[8] Другой ген, SMN2, считается геном, изменяющим заболевание, поскольку обычно чем больше SMN2 копий, тем легче течение болезни. Диагноз SMA основан на симптомах и подтверждается генетическое тестирование.[11][1]
Обычно мутация в SMN1 ген унаследованный от обоих родителей в аутосомно-рецессивный образом, хотя примерно в 2% случаев это происходит во время ранняя разработка (de novo ).[10][12] Частота спинальной мышечной атрофии во всем мире колеблется от примерно 1 на 4000 рождений до примерно 1 на 16000 рождений,[13] причем 1 из 7 000 и 1 из 10 000 обычно цитируются для Европы и США соответственно.[2]
Исход естественного течения болезни варьируется от нескольких месяцев в наиболее тяжелых случаях до нормальных. продолжительность жизни в более легких формах СМА.[8] Внедрение этих методов лечения в 2016 году значительно улучшило результаты. Лекарства, которые нацелены на генетическую причину заболевания, включают: Nusinersen, рисдиплам, и генная терапия медикамент онасемноген абепарвовец. Поддерживающая терапия включает физиотерапия, трудотерапия, респираторная поддержка, нутритивная поддержка, ортопедические вмешательства, и поддержка мобильности.[10]
Классификация
СМА проявляется в широком диапазоне степени тяжести, поражая младенцев и взрослых. Спектр заболеваний разделен на 3–5 типов в соответствии с наивысшей достигнутой вехой в моторном развитии.
Традиционная, наиболее часто используемая классификация выглядит следующим образом:
| Тип | Эпоним | Обычный возраст начала | Характеристики | OMIM |
|---|---|---|---|---|
| SMA 0 | Дородовой | Очень редкая форма, симптомы которой проявляются еще до рождения (снижение подвижности плода). У больных детей обычно есть только 1 копия SMN2 ген и обычно выживают всего несколько недель даже при интенсивной респираторной поддержке. | ||
| SMA 1 (Инфантильный) | Болезнь Верднига – Гофмана | 0–6 месяцев | Тяжелая форма проявляется в первые месяцы жизни, обычно с быстрым и неожиданным началом ("синдром гибкого ребенка "). Дети никогда не учатся сидеть без опоры. Быстрая смерть моторных нейронов приводит к неэффективности основных органов тела, особенно дыхательной системы. Дыхательная недостаточность, вызванная пневмонией, является наиболее частой причиной смерти. При отсутствии лечения и без респираторной поддержки у детей диагностируется СМА типа 1 обычно не доживает до двухлетнего возраста. Известно, что при надлежащей респираторной поддержке люди с более легкими фенотипами СМА типа 1, на которые приходится около 10% случаев СМА 1, доживают до подросткового и взрослого возраста. | 253300 |
| SMA 2 (Средний) | Болезнь Дубовица | 6–18 месяцев | Промежуточная форма поражает людей, которые были в состоянии хотя бы какое-то время в своей жизни сохранять сидячее положение, но никогда не учились ходить без опоры. Начало слабости обычно наблюдается в период между 6 и 18 месяцами жизни. Известно, что прогресс сильно различается: некоторые люди со временем постепенно ослабевают, в то время как другие при тщательном уходе остаются относительно стабильными. У этих детей обычно присутствует сколиоз, и его нужно исправить. ортез позвоночника, растущие стержни или же спондилодез может помочь улучшить дыхание. Мышцы тела ослаблены, и респираторная система вызывает серьезную озабоченность. Ожидаемая продолжительность жизни сокращается, но большинство людей со SMA 2 доживают до взрослого возраста. | 253550 |
| SMA 3 (Несовершеннолетний) | Болезнь Кугельберга-Веландера | > 12 месяцев | Юношеская форма обычно проявляется после 12 месяцев и описывает людей, которые могли ходить без поддержки хотя бы в течение некоторого времени в своей жизни, даже если позже они потеряли эту способность. Поражение дыхательных путей встречается реже, а ожидаемая продолжительность жизни нормальная или близкая к норме. Большинству людей с SMA 3 требуется поддержка мобильности. | 253400 |
| SMA 4 (Начало у взрослых) | Совершеннолетие | У взрослых форма (иногда классифицируемая как СМА типа 3 с поздним началом) обычно проявляется после третьего десятилетия жизни постепенным ослаблением мышц ног, что часто требует от человека использования вспомогательных приспособлений для ходьбы. Другие осложнения возникают редко, и это не влияет на ожидаемую продолжительность жизни. | 271150 |
В соответствии с новыми классификациями пациенты классифицируются на «не сидящих», «сидящих» и «ходящих» на основе их фактического функционального статуса.
Двигательное развитие и прогрессирование заболевания у людей с СМА обычно оценивается с использованием проверенных функциональных шкал - CHOP-INTEND (Тест нервно-мышечных заболеваний Детской больницы Филадельфии) или HINE (Хаммерсмитский неврологический осмотр новорожденных) у младенцев; и либо MFM (Motor Function Measure), либо один из нескольких вариантов HFMS (Hammersmith Functional Motor Scale)[14][15][16][17] у пожилых пациентов.
Одноименный лейбл Болезнь Верднига – Гофмана (иногда с ошибками с одним п) относится к самым ранним клиническим описаниям СМА у детей. Иоганн Хоффманн и Гвидо Вердниг. Одноименный термин Болезнь Кугельберга-Веландера после Эрик Клас Хендрик Кугельберг (1913–1983) и Лиза Веландер (1909–2001), которые отличили СМА от мышечной дистрофии.[18] Редко используемый Болезнь Дубовица (не путать с Синдром Дубовица ) назван в честь Виктор Дубовиц, английский невролог, автор нескольких исследований промежуточного фенотипа СМА.[нужна цитата ]
Признаки и симптомы

Симптомы различаются в зависимости от типа СМА, стадии заболевания, а также индивидуальных факторов. Приведенные ниже признаки и симптомы наиболее распространены при тяжелой СМА типа 0 / I:[19][требуется медицинская цитата ]
- Арефлексия, особенно в конечности
- Общий мышечная слабость, плохой мышечный тонус, вялость или склонность к провалу
- Проблемы с достижением вех в развитии, трудности при сидении / стоянии / ходьбе
- У маленьких детей: принятие положения лягушачьей ноги при сидении (бедра разведены, колени согнуты)
- Потеря прочности респираторный мышцы: слабые кашель, слабый крик (младенцы), скопление выделения в легких или горле, респираторный дистресс
- Колоколообразный торс (вызванный использованием только брюшных мышц для дыхания) при тяжелом типе СМА
- Обольщения (подергивание) языка
- Затрудненное сосание или глотание, плохое кормление
Причины

Спинальная мышечная атрофия связана с генетическая мутация в SMN1 ген.[20]
Человек хромосома 5 содержит два почти идентичных гена на место расположения 5q13: a теломерный копировать SMN1 и центромерный копировать SMN2. У здоровых людей SMN1 ген кодирует выживаемость двигательного нейрона белок (SMN), который, как следует из названия, играет решающую роль в выживании двигательные нейроны. В SMN2 ген, с другой стороны - из-за вариации в одном нуклеотид (840.C → T) - проходит альтернативное сращивание на стыке интрон 6 к экзон 8, только 10–20% SMN2 транскрипты, кодирующие полнофункциональную выживаемость двигательного нейрона белка (SMN-fl) и 80–90% транскриптов, в результате чего образуется усеченное белковое соединение (SMNΔ7), которое быстро разлагается в клетке.[21]
У людей, страдающих СМА, SMN1 ген мутировавший таким образом, что он не может правильно кодировать белок SMN - из-за либо удаление[22] происходящее в экзон 7[23] или другим точечные мутации (часто приводя к функциональному преобразованию SMN1 последовательность в SMN2). Однако почти у всех людей есть хотя бы одна функциональная копия SMN2 ген (у большинства из них 2–4), который все еще кодирует небольшое количество белка SMN - около 10–20% от нормального уровня, что позволяет некоторым нейронам выжить. Однако в долгосрочной перспективе снижение доступности белка SMN приводит к постепенной гибели клеток мотонейрона в передний рог спинного мозга и мозг. Мышцы, которые зависят от этих мотонейронов как нервные импульсы, теперь имеют пониженную иннервацию (также называемую денервация ), и, следовательно, снизились поступления от центральной нервной системы (ЦНС). Снижение передачи импульса через двигательные нейроны приводит к снижению сократительной активности денервированной мышцы. Следовательно, денервированные мышцы подвергаются прогрессирующей атрофия (чахнуть).[нужна цитата ]
Мышцы нижних конечности Обычно сначала поражаются мышцы верхних конечностей, позвоночника и шеи, а в более тяжелых случаях - легочные и жевательные мышцы. Проксимальный мышцы всегда поражаются раньше и в большей степени, чем дистальный.[24][нужна цитата ]
Тяжесть симптомов СМА во многом зависит от того, насколько хорошо сохраняются SMN2 гены могут восполнить потерю функции SMN1. Отчасти это связано с количеством SMN2 копии гена присутствуют на хромосоме. В то время как здоровые люди носят два SMN2 копий генов, люди с СМА могут иметь от 1 до 4 (или более) из них, причем большее количество SMN2 копии, тем легче степень тяжести заболевания. Таким образом, у большинства детей с СМА типа I есть один или два SMN2 копии; люди со СМА II и III обычно имеют не менее трех SMN2 копии; а у людей со SMA IV обычно их не менее четырех. Однако корреляция между тяжестью симптомов и SMN2 количество копий не является абсолютным, и, похоже, существуют другие факторы, влияющие на фенотип заболевания.[25]
Спинальная мышечная атрофия передается по наследству аутосомно-рецессивный паттерн, что означает, что дефектный ген расположен на аутосом. Для наследования заболевания необходимы две копии дефектного гена - по одной от каждого родителя: родители могут быть носителями и не затронуты лично. SMA вроде появляется de novo (т.е. без каких-либо наследственных причин) примерно в 2–4% случаев.
Спинальная мышечная атрофия поражает людей всех этнических групп, в отличие от других хорошо известных аутосомно-рецессивных заболеваний, таких как серповидноклеточная анемия и кистозный фиброз, которые имеют значительные различия в частоте встречаемости среди этнических групп. Общая распространенность SMA всех типов и среди всех этнических групп находится в пределах 1 на 10 000 человек; частота гена составляет примерно 1: 100, следовательно, примерно каждый 50 человек является носителем.[26][27] Нет известных последствий для здоровья носительства. Человек может узнать о статусе носителя только в том случае, если его ребенок страдает СМА или имеет SMN1 ген секвенирован.
Больные братья и сестры обычно имеют очень похожую форму СМА. Однако случаи различных типов СМА среди братьев и сестер действительно существуют - хотя и редко, эти случаи могут быть связаны с дополнительными de novo удаление SMN ген, не связанный с NAIP ген, или различия в SMN2 копировать числа.[нужна цитата ]
Диагностика
Наиболее тяжелые проявления в спектре СМА могут быть заметны для матери на поздних сроках беременности из-за уменьшения или отсутствия движений плода. Симптомы являются критическими (включая респираторный дистресс и плохое питание), которые обычно приводят к смерти в течение нескольких недель, в отличие от самого легкого фенотипа SMA (начало у взрослых), когда мышечная слабость может проявляться через десятилетия и прогрессировать до использования инвалидной коляски, но на всю жизнь ожидание без изменений.[28]
Наиболее частые клинические проявления спектра SMA, требующие диагностического генетического тестирования:
- Прогрессирующая двусторонняя мышечная слабость (как правило, в большей степени в верхней части рук и ног, чем в руках и ногах), которой предшествует бессимптомный период (все, кроме наиболее тяжелого типа 0)[28]
- Уплощение грудной стенки при вдохе и выпячивание живота при вдохе.
- гипотония связанные с отсутствиемрефлексы.
Хотя вышеперечисленные симптомы указывают на СМА, диагноз может быть подтвержден с абсолютной уверенностью только через генетическое тестирование для биаллельной делеции экзона 7 SMN1 ген, который является причиной более чем 95% случаев.[19] Генетическое тестирование обычно проводится с использованием образца крови и MLPA является одним из наиболее часто используемых методов генетического тестирования, так как он также позволяет установить количество SMN2 копии гена.[19]
Преимплантационное тестирование
Преимплантационная генетическая диагностика может использоваться для скрининга на наличие SMA эмбрионы в течение экстракорпоральное оплодотворение.
Пренатальное тестирование
Пренатальное тестирование для SMA возможно через биопсия хориона, внеклеточная ДНК плода анализ и другие методы.
Несущее тестирование
Те, кто рискуют оказаться перевозчики из SMN1 делеция и, следовательно, риск заражения потомства SMA, может пройти анализ на носитель с использованием образца крови или слюны. В Американский колледж акушеров и гинекологов рекомендует всем людям, которые думают о беременности, пройти тестирование, чтобы узнать, являются ли они носителями.[29] Частота носителя СМА сопоставима с другими заболеваниями, такими как талассемия, и в когорте северной Индии она составляет 1 из 38. [30] Однако генетическое тестирование не сможет выявить всех лиц, находящихся в группе риска, поскольку около 2% случаев вызваны: de novo мутации и 5% нормальных популяций имеют две копии SMN1 на одной хромосоме, что позволяет быть носителем, имея одну хромосому с двумя копиями и вторую хромосому с нулевыми копиями. Эта ситуация приведет к ложноотрицательный результат, поскольку статус носителя не будет правильно определен традиционным генетическим тестом.[31][32]
Скрининг новорожденных
Учитывая доступность методов лечения, которые кажутся наиболее эффективными на ранних стадиях заболевания, ряд экспертов рекомендуют регулярно проверять всех новорожденных детей на СМА.[33][34][35] В 2018 году скрининг новорожденных на SMA был добавлен в список рекомендуемых в США скрининговых тестов для новорожденных.[36][37][38] и по состоянию на май 2020 года он принят в 36 штатах США.[39] С 2020 года в Нидерландах обязателен скрининг новорожденных на СМА.[40] Кроме того, пилотные проекты по скринингу новорожденных на SMA были проведены в Австралии.[41] Бельгия,[42] Китай,[43] Германия,[44] Италия, Япония,[45] Тайвань,[46] и США.[47]
Управление
Лечение СМА зависит от степени тяжести и типа. В наиболее тяжелых формах (типы 0/1) люди имеют наибольшую мышечную слабость, требующую немедленного вмешательства. Принимая во внимание наименее тяжелую форму (тип 4 / начало у взрослых), люди могут не обращаться за определенными аспектами помощи до более позднего (десятилетия) в жизни. Хотя типы SMA и люди для каждого типа могут различаться, поэтому конкретные аспекты ухода за пациентами могут отличаться.[требуется медицинская цитата ]
Медикамент
Нусинерсен (Спинраза) используется для лечения мышечной атрофии позвоночника.[48] Это антисмысловой нуклеотид, который изменяет альтернативное сращивание из SMN2 ген.[48] Выдается непосредственно в Центральная нервная система используя интратекальная инъекция.[48][49] Нусинерсен продлевает выживаемость и улучшает двигательную функцию у младенцев со СМА.[50] [51] Он был одобрен для использования в США в 2016 году и для использования в ЕС в 2017 году.[52][53][54]
Онасемноген абепарвовец (Золгенсма) - это генная терапия лечение, которое использует самокомплементарный аденоассоциированный вирус типа 9 (scAAV-9) в качестве вектора для доставки SMN1 трансген.[55][56] Терапия была одобрена в США в 2019 году как внутривенный препарат для детей младше 24 месяцев.[57][58] В следующем году было получено разрешение в Европе и Японии.[59][60]
Рисдиплам (Evrysdi) - это лекарство, которое принимают устно в жидком виде.[61][62] Это пиридазин производная, которая работает за счет увеличения количества функциональных оставшийся в живых мотонейрон белок, производимый SMN2 ген через изменение схемы сращивания.[63][64] Рисдиплам был одобрен для медицинского применения в США в августе 2020 года.[61]
Дыхание
Дыхательная система является наиболее часто поражаемой системой, и осложнения являются основной причиной смерти при СМА типов 0/1 и 2. СМА типа 3 может иметь схожие респираторные проблемы, но они встречаются реже.[24] Осложнения, возникающие из-за ослабления межреберных мышц из-за отсутствия стимуляции со стороны нерва. Диафрагма поражается меньше, чем межреберные мышцы.[24] После ослабления мышцы никогда полностью не восстанавливают ту же функциональную способность, которая помогает при дыхании и кашле, а также других функциях. Таким образом, дыхание затруднено и создает риск недостаточного поступления кислорода / поверхностного дыхания и недостаточного очищения дыхательных путей. Эти проблемы чаще возникают во время сна, когда мышцы более расслаблены. Могут быть поражены глотательные мышцы глотки, что приводит к аспирации в сочетании с плохим механизмом кашля, что увеличивает вероятность заражения /пневмония.[65] Мобилизация и очистка секретов включает ручную или механическую физиотерапию грудной клетки с постуральным дренажом, а также ручное или механическое устройство для помощи при кашле. Чтобы помочь дыханию, Неинвазивная вентиляция (BiPAP ) часто используется и трахеостомия иногда может выполняться в более тяжелых случаях;[66] оба метода вентиляции продлевают выживаемость в сопоставимой степени, хотя трахеостомия препятствует развитию речи.[67]
Питание
Чем тяжелее тип СМА, тем выше вероятность возникновения проблем со здоровьем, связанных с питанием. Проблемы со здоровьем могут включать трудности с кормлением, открытием челюсти, жеванием и глотанием. Люди с такими трудностями могут подвергаться повышенному риску переедания или недоедания, недостаточного развития и стремления. Другие проблемы с питанием, особенно у неамбулаторных лиц (более тяжелые типы СМА), включают недостаточное прохождение пищи через желудок, желудочный рефлюкс, запор, рвоту и вздутие живота.[68][требуется медицинская цитата ] В связи с этим при СМА типа I и у людей с более тяжелым типом II может потребоваться питательная трубка или же гастростомия.[68][69][70] Кроме того, метаболические нарушения в результате нарушения СМА β-окисление из жирные кислоты в мышцах и может привести к органическая ацидемия и последующее повреждение мышц, особенно при голодании.[71][72] Предполагается, что люди со СМА, особенно с более тяжелыми формами заболевания, уменьшат потребление толстый и избегайте длительного голодания (т.е. ешьте чаще, чем здоровые люди)[73] а также выбирать более мягкую пищу, чтобы избежать аспирации.[65] Во время острого заболевания, особенно у детей, проблемы с питанием могут сначала проявиться или могут усугубить существующую проблему (например, аспирацию), а также вызвать другие проблемы со здоровьем, такие как нарушение электролитов и сахара в крови.[74][требуется медицинская цитата ]
Ортопедия
Проблемы со скелетом, связанные со слабостью мышц при СМА, включают тугие суставы с ограниченным диапазоном движений, вывихи бедра, деформацию позвоночника, остеопению, повышенный риск переломов и боли.[24] Слабые мышцы, которые обычно стабилизируют суставы, такие как позвоночник, приводят к развитию кифоз и / или сколиоз и совместная контрактура.[24] Спондилодез иногда выполняется у людей со СМА I / II, когда они достигают возраста 8–10 лет, чтобы уменьшить давление деформированного позвоночника на легкие. Кроме того, неподвижные люди, поза и положение на устройствах для передвижения, а также упражнения на диапазон движений и укрепление костей могут быть важны для предотвращения осложнений.[74] Людям с СМА также могут быть полезны различные формы физиотерапия, трудотерапия и физиотерапия.
Ортопедические приспособления могут использоваться для поддержки тела и облегчения ходьбы. Например, ортезы, такие как AFO (ортезы голеностопного сустава), используются для стабилизации стопы и для облегчения ходьбы, TLSO (грудные пояснично-крестцовые ортезы) используются для стабилизации торса. Вспомогательные технологии может помочь в управлении движением и повседневной активностью и значительно повысить качество жизни.
Другой
Хотя сердце не является предметом повседневного беспокойства, предполагается связь между СМА и некоторыми сердечными заболеваниями.[75][76][77][78]
Дети с СМА не отличаются от населения в целом по своему поведению; их когнитивное развитие могут быть немного быстрее, и некоторые аспекты их интеллект выше среднего.[79][80][81] Несмотря на свою инвалидность, люди, страдающие СМА, сообщают о высокой степени удовлетворения от жизни.[82]
Паллиативная помощь при СМА стандартизирована в Заявление о консенсусе в отношении стандартов медицинской помощи при спинальной мышечной атрофии[24] который был рекомендован для принятия во всем мире.
Прогноз
При отсутствии фармакологического лечения состояние людей со СМА со временем ухудшается. В последнее время увеличилась выживаемость у пациентов с тяжелой формой СМА при активной и активной поддерживающей респираторной и нутритивной поддержке.[83]
Если не лечить, большинство детей с диагнозом СМА типа 0 и я не достигают 4-летнего возраста, при этом повторяющиеся респираторные проблемы являются основной причиной смерти.[84] При надлежащем уходе более легкие случаи СМА типа I (на которые приходится примерно 10% всех случаев СМА1) доживают до взрослого возраста.[85] Долгосрочная выживаемость при СМА типа I недостаточно доказана; однако недавние достижения в области респираторной поддержки, похоже, снизили смертность.[86]
При нелеченой СМА типа II болезнь прогрессирует медленнее и продолжительность жизни меньше здорового населения. Часто случается смерть в возрасте до 20 лет, хотя многие люди с СМА доживают до родителей, бабушек и дедушек. СМА типа III имеет нормальную или почти нормальную продолжительность жизни при соблюдении стандартов медицинской помощи. Тип IV, СМА у взрослых обычно означает только нарушение подвижности и не влияет на продолжительность жизни.
Направления исследований
Поскольку основная генетическая причина СМА была выявлена в 1995 г.,[22] Было предложено и исследовано несколько терапевтических подходов, которые в первую очередь направлены на повышение доступности белка SMN в двигательных нейронах.[87] Основные направления исследований:
SMN1 замена гена
Генная терапия в SMA направлена на восстановление SMN1 функция гена за счет вставки специально созданных нуклеотид последовательность (a SMN1 трансген ) в ядро клетки используя вирусный вектор; scAAV -9 и scAAV-10 являются основными исследуемыми вирусными векторами. В 2019 году была одобрена терапия AAV9: Онасемноген абепарвовец.[88]
Только одна программа достигла клинической стадии. Работа по разработке генной терапии СМА также ведется в Институте миологии в Париже.[89] и на Оксфордский университет. В 2018 году также Биоген объявил о работе над генная терапия продукт для лечения SMA.[90]
SMN2 альтернативная модуляция сварки
Этот подход направлен на изменение альтернативное сращивание из SMN2 ген, чтобы заставить его кодировать более высокий процент полноразмерного белка SMN. Иногда это также называют генной конверсией, потому что она пытается преобразовать SMN2 ген функционально в SMN1 ген. Это терапевтический механизм одобренных препаратов нусинерсен и рисдиплам.
Дополнительный модулятор сплайсинга достиг клинической стадии разработки, а именно: бранаплам (LMI070, NVS-SM1), патентованный экспериментальный препарат на малых молекулах, вводимый перорально и разрабатываемый Новартис. По состоянию на октябрь 2017 г.[Обновить] соединение остается в фазе II клинических испытаний на младенцах с СМА типа 1, в то время как испытания на других категориях пациентов находятся в стадии разработки.[91]
Из прекращенных молекул на клинической стадии RG3039, также известный как хиназолин495, был запатентованным хиназолин производная, разработанная Repligen и имеет лицензию на Pfizer в марте 2014 года, который был прекращен вскоре после того, как завершились только испытания фазы I. PTK-SMA1 был запатентованным модулятором сплайсинга малых молекул тетрациклины группа, разработанная Paratek Pharmaceuticals и собирающаяся начать клиническую разработку в 2010 году, чего, однако, так и не произошло. RG7800 - это молекула, родственная RG7916, разработанная Hoffmann-La Roche, которая прошла фазу I.[92]
Фундаментальные исследования также выявили другие соединения, которые модифицировали SMN2 сращивание in vitro, подобно ортованадат натрия[93] и акларубицин.[94] Морфолино Антисмысловые олигонуклеотиды -типа с той же клеточной мишенью, что и нузинерсен, остаются предметом интенсивных исследований, в том числе в Университетский колледж Лондона[95] и на Оксфордский университет.[96]
SMN2 активация гена
Этот подход направлен на повышение экспрессии (активности) SMN2 ген, тем самым увеличивая количество доступного полноразмерного белка SMN.
- Устный сальбутамол (альбутерол), популярный астма медицины, показал терапевтический потенциал при СМА как in vitro[97] и в трех небольших клинических испытаниях с участием пациентов со СМА 2 и 3 типа,[98][99][100] помимо респираторных преимуществ.
Несколько соединений изначально были многообещающими, но не смогли продемонстрировать эффективность в клинических испытаниях:
- Бутираты (бутират натрия и фенилбутират натрия ) обещал в in vitro исследования[101][102][103] но клинические испытания на людях с симптомами не подтвердили их эффективность.[104] Еще одно клиническое испытание на младенцах с предсимптоматическим типом 1–2 было завершено в 2015 г., но результаты не были опубликованы.[105]
- Вальпроевая кислота (VPA) использовался в SMA на экспериментальной основе в 1990-х и 2000-х годах, потому что in vitro исследования показали его умеренную эффективность.[106][107] Однако он не продемонстрировал эффективности в достижимых концентрациях при проведении крупных клинических испытаний.[108][109][110] Также было высказано предположение, что он может быть эффективным в подгруппе людей с СМА, но его действие может подавляться транслоказа жирных кислот в других.[111] Другие утверждают, что на самом деле это может усугубить симптомы СМА.[112] В настоящее время он не используется из-за риска серьезных побочных эффектов, связанных с длительным использованием. Мета-анализ 2019 года показал, что VPA может предложить преимущества даже без повышения функциональной оценки.[113]
- Гидроксикарбамид (гидроксимочевина) эффективна на моделях мышей[114] и впоследствии коммерческие исследования Ново Нордиск, Дания, но в последующих клинических испытаниях не продемонстрировал никакого эффекта у людей со СМА.[115]
Соединения, которые увеличили SMN2 Мероприятия in vitro но не дошло до клинической стадии включать гормон роста, разные ингибиторы гистондеацетилазы,[116] бензамид M344,[117] гидроксамовые кислоты (CBHA, SBHA, энтиностат, панобиностат,[118] трихостатин А,[119][120] вориностат[121]), пролактин[122] а также натуральный полифенол соединения, подобные ресвератрол и куркумин.[123][124] Целекоксиб, а путь p38 активатор, иногда используется людьми с СМА не по назначению на основании исследования на одном животном[125] но такое использование не подтверждено исследованиями на клинической стадии.
Стабилизация SMN
Стабилизация SMN направлена на стабилизацию белка SMNΔ7, короткоживущего дефектного белка, кодируемого SMN2 ген, так что он способен поддерживать нервные клетки.[126]
Никакие соединения не были переведены на клиническую стадию. Аминогликозиды показали способность увеличивать доступность белка SMN в двух исследованиях.[127][128] Индопрофен предложил некоторое обещание in vitro.[129]
Нейропротекция
Нейропротекторный лекарства направлены на обеспечение выживания мотонейронов даже при низком уровне белка SMN.
- Олезоксим запатентованный нейропротекторный состав, разработанный французской компанией Трофос, позже приобретенный Hoffmann-La Roche, который показал стабилизирующий эффект в клинических испытаниях фазы II с участием людей со СМА 2 и 3 типа. Его разработка была прекращена в 2018 году ввиду конкуренции с Спинраза и хуже, чем ожидалось, данные, полученные в результате открытого исследования расширения.[130]
Из клинически изученных соединений, не проявивших эффективности, тиреотропин-рилизинг-гормон (TRH) многообещал в Открой надпись неконтролируемый клиническое испытание[131][132][133] но не оказался эффективным в последующем двойной слепой плацебо-контролируемый испытание.[134] Рилузол, препарат, имеющий умеренную клиническую пользу при боковой амиотрофический склероз, было предложено аналогичным образом протестировать в SMA,[135][136] однако исследование 2008–2010 гг. при СМА 2 и 3 типа[137] был прекращен досрочно из-за отсутствия удовлетворительных результатов.[138]
Соединения, оказавшие нейропротекторное действие на in vitro исследования, но никогда не переезжал in vivo исследования включают β-лактамные антибиотики (например., цефтриаксон )[139][140] и фоллистатин.[141]
Восстановление мышц
Этот подход направлен на противодействие эффекту SMA путем воздействия на мышечную ткань, а не на нейроны.
- СК-2127107 (СК-107) - скелетный тропонин активатор, разработанный Cytokinetics в сотрудничестве с Астеллас. Препарат направлен на увеличение мышечной реактивности, несмотря на снижение нейронной передачи сигналов. По состоянию на октябрь 2016 г.[Обновить]молекула проходит II фазу клинических испытаний у подростков и взрослых со СМА 2, 3 и 4 типа.[142]
Стволовые клетки
В 2013–2014 гг. Небольшое количество детей с СМА1 в Италии получили инъекции стволовых клеток по решению суда после Мошенничество с выносливостью, но сообщалось, что лечение не дало эффекта.[143][144]
Хотя стволовые клетки никогда не являются частью какой-либо признанной терапии СМА, ряд частных компаний, обычно расположенных в странах со слабым регуляторным надзором, используют преимущества шумиха в СМИ и продавать инъекции стволовых клеток как «лекарство» от широкого спектра заболеваний, включая СМА. Медицинский консенсус состоит в том, что такие процедуры не приносят клинической пользы, хотя и сопряжены со значительным риском, поэтому людям с СМА их не рекомендуется.[145][146]
Реестры
Люди с СМА в Евросоюз могут участвовать в клинических исследованиях, вводя свои данные в реестры, управляемые TREAT-NMD.[147]
Смотрите также
Рекомендации
- ^ а б c d «Спинальная мышечная атрофия». Информационный центр по генетическим и редким заболеваниям (GARD) - программа NCATS. Получено 27 мая 2019.
- ^ а б c d е ж грамм «Спинальная мышечная атрофия». NORD (Национальная организация по редким заболеваниям). Получено 27 мая 2019.
- ^ «Спинальная мышечная атрофия». nhs.uk. 23 октября 2017 г.. Получено 24 октября 2020.
- ^ а б «Спинальная мышечная атрофия: MedlinePlus Genetics». medlineplus.gov. Получено 24 октября 2020.
- ^ "Спинальная мышечная атрофия (SMA) | Бостонская детская больница". www.childrenshospital.org. Получено 25 октября 2020.
- ^ «FDA одобряет инновационную генную терапию для лечения педиатрических пациентов со спинальной мышечной атрофией, редким заболеванием и ведущей генетической причиной детской смертности». FDA. 24 мая 2019. Получено 27 мая 2019.
- ^ "Информационный бюллетень о спинальной мышечной атрофии | Национальный институт неврологических заболеваний и инсульта". NINDS. Получено 27 мая 2019.
- ^ а б c d е «Спинальная мышечная атрофия». Домашний справочник по генетике. Получено 27 мая 2019.
- ^ а б «Спинальная мышечная атрофия». NORD (Национальная организация по редким заболеваниям). Получено 27 мая 2019.
- ^ а б c «Спинальная мышечная атрофия». Информационный центр по генетическим и редким заболеваниям (GARD) - программа NCATS. Получено 27 мая 2019.
- ^ "Спинальная мышечная атрофия - Условия | Детский национальный". childrensnational.org. Получено 25 октября 2020.
- ^ Prior, Thomas W .; Leach, Meganne E .; Finanger, Erika (1993), Adam, Margaret P .; Ardinger, Holly H .; Пагон, Роберта А .; Уоллес, Стефани Э. (ред.), «Спинальная мышечная атрофия», GeneReviews®, Сиэтл (Вашингтон): Вашингтонский университет, Сиэтл, PMID 20301526, получено 25 октября 2020
- ^ Verhaart, Ingrid E.C .; Робертсон, Агата; Лири, Ребекка; Макмакен, Грейс; Кениг, Кирстен; Киршнер, Янбернд; Джонс, Синтия С .; Повар, Сюзанна Ф .; Лохмюллер, Ханс (июль 2017 г.). «Подход из нескольких источников для определения заболеваемости СМА и населения, готового к исследованиям». Журнал неврологии. 264 (7): 1465–1473. Дои:10.1007 / s00415-017-8549-1. ISSN 0340-5354. ЧВК 5502065. PMID 28634652.
- ^ Мэйн М, Кайрон Х, Меркури Э, Мунтони Ф (2003). «Функциональная моторная шкала Hammersmith для детей со спинальной мышечной атрофией: шкала для проверки способностей и отслеживания прогресса у детей с ограниченными возможностями передвижения». Европейский журнал детской неврологии. 7 (4): 155–9. Дои:10.1016 / S1090-3798 (03) 00060-6. PMID 12865054.
- ^ Krosschell KJ, Maczulski JA, Crawford TO, Scott C, Swoboda KJ (июль 2006 г.). «Модифицированная функциональная моторная шкала Хаммерсмита для использования в многоцентровых исследованиях спинальной мышечной атрофии». Нервно-мышечные расстройства. 16 (7): 417–26. Дои:10.1016 / j.nmd.2006.03.015. ЧВК 3260054. PMID 16750368.
- ^ О'Хаген Дж. М., Гланцман А. М., Макдермотт М. П., Райан П. А., Фликингер Дж., Куигли Дж., Райли С., Санборн Э., Ирвин С., Мартенс В. Б., Аннис С., Тавил Р., Оскуи М., Даррас Б. Т., Финкель Р. С., Де Виво, округ Колумбия (Октябрь 2007 г.). «Расширенная версия функциональной моторной шкалы Hammersmith для пациентов со СМА II и III». Нервно-мышечные расстройства. 17 (9–10): 693–7. Дои:10.1016 / j.nmd.2007.05.009. PMID 17658255. S2CID 10365924.
- ^ Гланцман А.М., О'Хаген Дж. М., Макдермотт М. П., Мартенс В. Б., Фликингер Дж., Райли С., Куигли Дж., Монтес Дж., Данауэй С., Дэн Л., Чанг В. К., Тавил Р., Даррас Б. Т., Де Виво Д. К., Кауфманн П., Финкель Р. С. , и другие. (Сеть педиатрических нейромышечных клинических исследований спинальной мышечной атрофии (PNCR)) (декабрь 2011 г.). «Валидация расширенной функциональной моторной шкалы Хаммерсмита при спинальной мышечной атрофии II и III типов». Журнал детской неврологии. 26 (12): 1499–507. Дои:10.1177/0883073811420294. PMID 21940700. S2CID 206549483.
- ^ Дубовиц V (январь 2009 г.). «Бред в истории спинальной мышечной атрофии». Нервно-мышечные расстройства. 19 (1): 69–73. Дои:10.1016 / j.nmd.2008.10.004. PMID 18951794. S2CID 37576912.
- ^ а б c Оскуи М., Даррас Б.Т., ДеВиво, округ Колумбия (2017). "Глава 1". В Sumner CJ, Paushkin S, Ko CP (ред.). Спинальная мышечная атрофия: механизмы заболевания. Эльзевир. ISBN 978-0-12-803685-3.
- ^ Бжустович Л.М., Ленер Т., Кастилья Л.Х., Пенчасзаде Г.К., Вильгельмсен К.С., Дэниэлс Р., Дэвис К.Э., Лепперт М., Зитер Ф., Вуд Д. (апрель 1990 г.). «Генетическое картирование хронической мышечной атрофии позвоночника в детстве по хромосоме 5q11.2-13.3». Природа. 344 (6266): 540–1. Bibcode:1990Натура.344..540Б. Дои:10.1038 / 344540a0. PMID 2320125. S2CID 4259327.
- ^ «Спинальная мышечная атрофия». Домашний справочник по генетике. Получено 15 мая 2019.
- ^ а б Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M (январь 1995 г.). «Идентификация и характеристика гена, определяющего мышечную атрофию позвоночника». Клетка. 80 (1): 155–65. Дои:10.1016/0092-8674(95)90460-3. PMID 7813012. S2CID 14291056.
- ^ Пассини М.А., Бу Дж., Ричардс А.М., Киннеком С., Сарди С.П., Станек Л.М., Хуа И., Риго Ф., Матсон Дж., Хунг Дж., Кай Е.М., Шихабуддин Л.С., Крайнер А.Р., Беннетт К.Ф., Ченг С.Х. «Антисмысловые олигонуклеотиды, доставленные в ЦНС мыши, уменьшают симптомы тяжелой спинальной мышечной атрофии». Научная трансляционная медицина. 3 (72): 72ра18. Дои:10.1126 / scitranslmed.3001777. ЧВК 3140425. PMID 21368223.
- ^ а б c d е ж Ван Ч., Финкель Р.С., Бертини Э.С., Шрот М., Саймондс А., Вонг Б., Алоизиус А., Моррисон Л., Мейн М., Кроуфорд ТО, Трела А. (август 2007 г.). «Заявление о консенсусе по стандарту лечения спинальной мышечной атрофии». Журнал детской неврологии. 22 (8): 1027–49. Дои:10.1177/0883073807305788. PMID 17761659. S2CID 6478040.
- ^ Jedrzejowska M, Milewski M, Zimowski J, Borkowska J, Kostera-Pruszczyk A, Sielska D, Jurek M, Hausmanowa-Petrusewicz I (2009). «Модификаторы фенотипа спинальной мышечной атрофии: количество копий гена SMN2, делеция в гене NAIP и, возможно, пол влияют на течение заболевания». Acta Biochimica Polonica. 56 (1): 103–8. Дои:10.18388 / abp.2009_2521. PMID 19287802.
- ^ Су Й.Н., Хунг С.К., Лин С.И., Чен Ф.Й., Черн Ю.П., Цай С., Чанг Т.С., Ян С.К., Ли Х., Хо Х.Н., Ли С.Н. Schrijver I (ред.). «Скрининг носителей на предмет спинальной мышечной атрофии (СМА) у 107 611 беременных женщин в период 2005–2009 годов: проспективное популяционное когортное исследование». PLOS ONE. 6 (2): e17067. Bibcode:2011PLoSO ... 617067S. Дои:10.1371 / journal.pone.0017067. ЧВК 3045421. PMID 21364876.
- ^ Шугарман Э.А., Наган Н., Чжу Х., Акмаев В.Р., Чжоу З., Рольфс Э.М., Флинн К., Хендриксон BC, Шолл Т., Сирко-Осадса Д.А., Аллитто Б.А. (январь 2012 г.). «Панэтнический скрининг носителей и пренатальная диагностика спинальной мышечной атрофии: клинический лабораторный анализ> 72 400 образцов». Европейский журнал генетики человека. 20 (1): 27–32. Дои:10.1038 / ejhg.2011.134. ЧВК 3234503. PMID 21811307.
- ^ а б Ottesen EW (январь 2017 г.). «ISS-N1 является первым одобренным FDA лекарством от спинальной мышечной атрофии». Трансляционная нейробиология. 8 (1): 1–6. Дои:10.1515 / tnsci-2017-0001. ЧВК 5382937. PMID 28400976.
- ^ «Скрининг носителей в эпоху геномной медицины - ACOG». www.acog.org. Получено 24 февраля 2017.
- ^ Нилай, М., Мойрантхем, А., Саксена, Д., Мандал, К., Пхадке, С.Р. (октябрь 2020 г.). «Частота носителей спинальной мышечной атрофии, связанной с SMN1, у населения Северной Индии: необходимость в программе скрининга на уровне населения». Американский журнал медицинской генетики, часть A: 1–4. Дои:10.1002 / ajmg.a.61918.
- ^ Prior TW (ноябрь 2008 г.). «Скрининг носителей при спинальной мышечной атрофии». Генетика в медицине. 10 (11): 840–2. Дои:10.1097 / GIM.0b013e318188d069. ЧВК 3110347. PMID 18941424.
- ^ Ar Rochmah M, Awano H, Awaya T, Harahap NI, Morisada N, Bouike Y, Saito T, Kubo Y, Saito K, Lai PS, Morioka I, Iijima K, Nishio H, Shinohara M (ноябрь 2017 г.). «Носители спинальной мышечной атрофии с двумя копиями SMN1». Мозг и развитие. 39 (10): 851–860. Дои:10.1016 / j.braindev.2017.06.002. PMID 28676237. S2CID 26504674.
- ^ Серра-Джухе С., Тиццано Э. Ф. (декабрь 2019 г.). «Перспективы генетического консультирования при спинальной мышечной атрофии в новую терапевтическую эру: раннее предсимптоматическое вмешательство и тестирование у несовершеннолетних». Европейский журнал генетики человека. 27 (12): 1774–1782. Дои:10.1038 / s41431-019-0415-4. ЧВК 6871529. PMID 31053787.
- ^ Гласкок Дж., Сэмпсон Дж., Хайдет-Филлипс А., Коннолли А., Даррас Б., Дэй Дж и др. (29 мая 2018 г.). «Алгоритм лечения младенцев, у которых диагностирована спинальная мышечная атрофия посредством скрининга новорожденных». Журнал нервно-мышечных заболеваний. 5 (2): 145–158. Дои:10.3233 / JND-180304. ЧВК 6004919. PMID 29614695.
- ^ Dangouloff T, Burghes A, Tizzano EF, Servais L (январь 2020 г.). «244-й международный семинар ENMC: Скрининг новорожденных на спинальную мышечную атрофию 10–12 мая 2019 г., Хофдорп, Нидерланды». Нервно-мышечные расстройства. 30 (1): 93–103. Дои:10.1016 / j.nmd.2019.11.002. PMID 31882184.
- ^ Лопес Дж. М. (16 июля 2018 г.). «SMA добавлена в список рекомендуемых скринингов на заболевания, назначенные ...» Новости SMA сегодня. Получено 4 мая 2020.
- ^ Стивенсон К. (5 июля 2018 г.). «SMA добавлена в национальный список заболеваний, которые необходимо проверять при рождении». Ассоциация мышечной дистрофии. Получено 4 мая 2020.
- ^ «Рекомендуемая единообразная экранирующая панель». Официальный веб-сайт Управления ресурсов и служб здравоохранения США. 3 июля 2017 г.. Получено 4 мая 2020.
- ^ Макколл С. «Скрининг новорожденных на спинальную мышечную атрофию». Вылечить SMA. Получено 4 мая 2020.
- ^ Министр ван Фольксгезондхайд, Welzijn en Sport (23 июля 2019 г.). «Неонатальный скрининг на спинальную мышечную атрофию - Консультативный отчет - Совет здравоохранения Нидерландов». www.healthcouncil.nl. Получено 4 мая 2020.
- ^ Кариявасам Д.С., Рассел Дж. С., Вайли В., Александр И. Е., Фаррар М. А. (март 2020 г.). «Проведение скрининга новорожденных на спинальную мышечную атрофию: опыт Австралии». Генетика в медицине. 22 (3): 557–565. Дои:10.1038 / s41436-019-0673-0. PMID 31607747. S2CID 204459317.
- ^ Бомер Ф., Каберг Дж. Х., Дидеберг В., Дарденн Д., Бурс В., Хилигсманн М. и др. (Май 2019). «Скрининг новорожденных на SMA в Южной Бельгии». Нервно-мышечные расстройства. 29 (5): 343–349. Дои:10.1016 / j.nmd.2019.02.003. PMID 31030938. S2CID 72332212.
- ^ Lin Y, Lin CH, Yin X, Zhu L, Yang J, Shen Y и др. (2019). «Скрининг новорожденных на спинальную мышечную атрофию в Китае с использованием масс-спектрометрии ДНК». Границы генетики. 10: 1255. Дои:10.3389 / fgene.2019.01255. ЧВК 6928056. PMID 31921298.
- ^ Вилл К., Кельбель Х., Шварц О, Блашек А., Ольгемеллер Б., Хармс Э. и др. (31 октября 2019 г.). «Годовое обследование новорожденных на СМА - результаты немецкого пилотного проекта». Журнал нервно-мышечных заболеваний. 6 (4): 503–515. Дои:10.3233 / JND-190428. ЧВК 6918901. PMID 31594245.
- ^ Шинохара М., Ниба И.Т., Виджая Ю.О., Такаяма И., Мицуиси К., Кумасака С., Кондо Ю., Такатера А., Хокуто И., Мориока И., Огивара К. (декабрь 2019 г.). «Новая система для скрининга спинальной мышечной атрофии у новорожденных: японское пилотное исследование». Международный журнал неонатального скрининга. 5 (4): 41. Дои:10.3390 / ijns5040041.
- ^ Chien YH, Chiang SC, Weng WC, Lee NC, Lin CJ, Hsieh WS и др. (Ноябрь 2017 г.). «Пресимптоматическая диагностика спинальной мышечной атрофии посредством скрининга новорожденных». Журнал педиатрии. 190: 124–129.e1. Дои:10.1016 / j.jpeds.2017.06.042. PMID 28711173. S2CID 20621772.
- ^ Kraszewski JN, Kay DM, Stevens CF, Koval C, Haser B, Ortiz V, et al. (Июнь 2018). «Пилотное исследование популяционного скрининга новорожденных на спинальную мышечную атрофию в штате Нью-Йорк». Генетика в медицине. 20 (6): 608–613. Дои:10.1038 / gim.2017.152. PMID 29758563.
- ^ а б c «Спинразанусинерсен для инъекций, раствор». DailyMed. 30 июнь 2020. Получено 8 августа 2020.
- ^ Грант C (27 декабря 2016 г.). "Неожиданное одобрение лекарств - это праздничный подарок для Biogen". Журнал "Уолл Стрит. ISSN 0099-9660. Получено 27 декабря 2016.
- ^ Финкель Р.С., Меркури Э., Даррас Б.Т., Коннолли А.М., Кунц Н.Л., Киршнер Дж. И др. (Ноябрь 2017 г.). «Нусинерсен против фиктивного контроля при спинальной мышечной атрофии с младенческим началом». Медицинский журнал Новой Англии. 377 (18): 1723–32. Дои:10.1056 / NEJMoa1702752. PMID 29091570. S2CID 4771819.
- ^ Wadman, Renske I .; ван дер Поль, В. Людо; Bosboom, Венди Mj; Ассельман, Фэй-Линн; ван ден Берг, Леонард Х .; Iannaccone, Susan T .; Вранкен, Александр Фье (1 июня 2020 г.). «Медикаментозное лечение мышечной атрофии позвоночника II и III типов». Кокрановская база данных систематических обзоров. 1: CD006282. Дои:10.1002 / 14651858.CD006282.pub5. ISSN 1469-493X. ЧВК 6995983. PMID 32006461.
- ^ «Спинраза (нусинерсен) для инъекций». НАС. Управление по контролю за продуктами и лекарствами (FDA). 18 января 2017 г.. Получено 8 августа 2020.
- ^ «Спинраза EPAR». Европейское агентство по лекарствам (EMA). Получено 8 августа 2020.
- ^ «Спинраза (Нусинерсен) одобрена в Европейском Союзе как первое средство для лечения спинальной мышечной атрофии». Агентство Франс-Пресс (AFP). 1 июня 2017 г.. Получено 1 июня 2017.
- ^ «Золгенсма 2 x 1013 векторных геномов / мл раствор для инфузии». www.medicines.org.uk. Получено 8 августа 2020.
- ^ «Золгенсма-онасемноген абепарвовец-xioi комплект». DailyMed. 24 мая 2019. Получено 8 августа 2020.
- ^ «FDA одобряет инновационную генную терапию для лечения педиатрических пациентов со спинальной мышечной атрофией, редким заболеванием и ведущей генетической причиной детской смертности». НАС. Управление по контролю за продуктами и лекарствами (FDA) (Пресс-релиз). 24 мая 2019. Получено 27 мая 2019.
 Эта статья включает текст из этого источника, который находится в всеобщее достояние.
Эта статья включает текст из этого источника, который находится в всеобщее достояние. - ^ «Золгенсма». НАС. Управление по контролю за продуктами и лекарствами (FDA). 24 мая 2019. Получено 8 августа 2020.
- ^ «Золгенсма ЕПАР». Европейское агентство по лекарствам (EMA). 24 марта 2020 г.. Получено 8 августа 2020.
- ^ «Новартис получил одобрение Министерства здравоохранения, труда и социального обеспечения Японии на Zolgensma - единственную генную терапию для пациентов со спинальной мышечной атрофией (СМА)». Новартис (Пресс-релиз). Получено 8 августа 2020.
- ^ а б «FDA одобряет пероральное лечение спинальной мышечной атрофии». НАС. Управление по контролю за продуктами и лекарствами (FDA) (Пресс-релиз). 7 августа 2020 г.. Получено 7 августа 2020.
- ^ «Эврисди (рисдиплам) для раствора для приема внутрь» (PDF). Genentech. Получено 8 августа 2020.
- ^ Мария Жоао Алмейда (8 сентября 2016 г.). "RG7916". Услуги BioNews. Получено 8 октября 2017.
- ^ Чжао X, Фэн З., Линг К.К., Моллин А., Шиди Дж., Йе С. и др. (Май 2016). «Фармакокинетика, фармакодинамика и эффективность низкомолекулярного модификатора сплайсинга SMN2 на мышиных моделях спинальной мышечной атрофии». Молекулярная генетика человека. 25 (10): 1885–1899. Дои:10.1093 / hmg / ddw062. ЧВК 5062580. PMID 26931466.
- ^ а б Бодамер О. (ноябрь 2017 г.). «Спинальная мышечная атрофия». uptodate.com. Получено 1 декабря 2017.
- ^ Бах-младший, Ниранджан В., Уивер Б. (апрель 2000 г.). «Спинальная мышечная атрофия 1 типа: неинвазивный подход к лечению дыхательных путей». Грудь. 117 (4): 1100–5. Дои:10.1378 / сундук.117.4.1100. PMID 10767247.
- ^ Бах JR, Saltstein K, Sinquee D, Weaver B, Komaroff E (май 2007). «Долгосрочная выживаемость при болезни Верднига-Гофмана». Американский журнал физической медицины и реабилитации. 86 (5): 339–45 quiz 346–8, 379. Дои:10.1097 / PHM.0b013e31804a8505. PMID 17449977. S2CID 9942245.
- ^ а б Messina S, Pane M, De Rose P, Vasta I, Sorleti D, Aloysius A, Sciarra F, Mangiola F, Kinali M, Bertini E, Mercuri E (май 2008 г.). «Проблемы с питанием и недостаточность питания при спинальной мышечной атрофии II типа». Нервно-мышечные расстройства. 18 (5): 389–93. Дои:10.1016 / j.nmd.2008.02.008. PMID 18420410. S2CID 23302291.
- ^ Чен Ю.С., Ши Х.Х., Чен Т.Х., Куо СН, Чон Й.Дж. (март 2012 г.) «Распространенность и факторы риска затруднений кормления и глотания при спинальной мышечной атрофии II и III типов». Журнал педиатрии. 160 (3): 447–451.e1. Дои:10.1016 / j.jpeds.2011.08.016. PMID 21924737.
- ^ Тилтон А.Х., Миллер, доктор медицины, Хошу В. (июнь 1998 г.). «Питание и глотание у педиатрических нервно-мышечных больных». Семинары по детской неврологии. 5 (2): 106–15. Дои:10.1016 / S1071-9091 (98) 80026-0. PMID 9661244.
- ^ Tein I, Sloane AE, Доннер EJ, Lehotay DC, Millington DS, Kelley RI (январь 1995 г.). «Нарушения окисления жирных кислот при спинальной мышечной атрофии в детстве: первичный или вторичный дефект (ы)?». Детская неврология. 12 (1): 21–30. Дои:10.1016 / 0887-8994 (94) 00100-Г. PMID 7748356.
- ^ Кроуфорд ТО, Сладки Дж. Т., Хурко О., Беснер-Джонстон А., Келли Р. И. (март 1999 г.). «Аномальный метаболизм жирных кислот при мышечной атрофии позвоночника у детей». Анналы неврологии. 45 (3): 337–43. Дои:10.1002 / 1531-8249 (199903) 45: 3 <337 :: AID-ANA9> 3.0.CO; 2-U. PMID 10072048.
- ^ Лейтон С (2003). «Проблемы питания, связанные с мышечной атрофией позвоночника». Питание и диетология. 60 (2): 92–96.
- ^ а б Apkon S (лето 2017 г.). "SMA CARE SERIES - Опорно-двигательная система" (PDF). www.curesma.org.
- ^ Рудник-Шёнеборн С., Хеллер Р., Берг С., Бецлер С., Гримм Т., Эггерманн Т., Эггерманн К., Вирт Р., Вирт Б., Зеррес К. (октябрь 2008 г.). «Врожденный порок сердца - это признак тяжелой детской спинальной мышечной атрофии». Журнал медицинской генетики. 45 (10): 635–8. Дои:10.1136 / jmg.2008.057950. PMID 18662980. S2CID 7170069.
- ^ Heier CR, Satta R, Lutz C, DiDonato CJ (октябрь 2010 г.). «Аритмия и сердечные дефекты являются особенностями мышей, моделирующих мышечную атрофию позвоночника». Молекулярная генетика человека. 19 (20): 3906–18. Дои:10,1093 / hmg / ddq330. ЧВК 2947406. PMID 20693262.
- ^ Шабаби М., Хабиби Дж., Ян Х.Т., Вейл С.М., Сьюэлл В.А., Лорсон С.Л. (октябрь 2010 г.). «Пороки сердца вносят вклад в патологию моделей спинальной мышечной атрофии». Молекулярная генетика человека. 19 (20): 4059–71. Дои:10.1093 / hmg / ddq329. PMID 20696672.
- ^ Беван А.К., Хатчинсон К.Р., Фуст К.Д., Браун Л., Макговерн В.Л., Шмельцер Л., Уорд Дж.Г., Петруска Дж.С., Луччеси П.А., Бургес А.Х., Каспар Б.К. (октябрь 2010 г.). «Ранняя сердечная недостаточность в модели спинальной мышечной атрофии SMNDelta7 и коррекция постнатальной доставки scAAV9-SMN». Молекулярная генетика человека. 19 (20): 3895–905. Дои:10,1093 / hmg / ddq300. ЧВК 2947399. PMID 20639395.
- ^ фон Гонтард А., Зеррес К., Бакес М., Лауферсвайлер-Пласс С., Вендланд С., Мельхерс П., Лемкуль Г., Рудник-Шёнеборн С. (февраль 2002 г.). «Интеллект и когнитивные функции у детей и подростков со спинальной мышечной атрофией». Нервно-мышечные расстройства. 12 (2): 130–6. Дои:10.1016 / S0960-8966 (01) 00274-7. PMID 11738354. S2CID 46694209.
- ^ Billard C, Gillet P, Signoret JL, Uicaut E, Bertrand P, Fardeau M, Barthez-Carpentier MA, Santini JJ (1992). «Когнитивные функции при мышечной дистрофии Дюшенна: переоценка и сравнение со спинальной мышечной атрофией». Нервно-мышечные расстройства. 2 (5–6): 371–8. Дои:10.1016 / S0960-8966 (06) 80008-8. PMID 1300185. S2CID 22211725.
- ^ Laufersweiler-Plass C, Rudnik-Schöneborn S, Zerres K, Backes M, Lehmkuhl G, von Gontard A (январь 2003 г.). «Поведенческие проблемы у детей и подростков со спинальной мышечной атрофией и их братьев и сестер». Медицина развития и детская неврология. 45 (1): 44–9. Дои:10.1017 / S0012162203000082. PMID 12549754.
- ^ де Оливейра CM, Араужу AP (январь 2011 г.). «Качество жизни по самооценке не коррелирует с функциональным статусом у детей и подростков со спинальной мышечной атрофией». Европейский журнал детской неврологии. 15 (1): 36–9. Дои:10.1016 / j.ejpn.2010.07.003. PMID 20800519.
- ^ Даррас Б., Финкель Р. (2017). Спинальная мышечная атрофия. Великобритания, США: Эльзевир. п. 417. ISBN 978-0-12-803685-3.
- ^ Юань Н., Ван Ч., Трела А., Альбанезе, CT (июнь 2007 г.). «Лапароскопическая фундопликация Ниссена во время установки гастростомической трубки и неинвазивной вентиляции может улучшить выживаемость при спинальной мышечной атрофии I и тяжелой степени II». Журнал детской неврологии. 22 (6): 727–31. Дои:10.1177/0883073807304009. PMID 17641258. S2CID 38799022.
- ^ Бах-младший (май 2007 г.). «Медицинские аспекты долговременной выживаемости при болезни Верднига-Гофмана». Американский журнал физической медицины и реабилитации. 86 (5): 349–55. Дои:10.1097 / PHM.0b013e31804b1d66. PMID 17449979. S2CID 39989993.
- ^ Оскуи М., Леви Дж., Гарланд Си-Джей, Грей Дж. М., О'Хаген Дж., Де Виво, округ Колумбия, Кауфманн П. (ноябрь 2007 г.). «Изменяющееся естествознание спинальной мышечной атрофии 1 типа». Неврология. 69 (20): 1931–6. Дои:10.1212 / 01.wnl.0000290830.40544.b9. PMID 17998484. S2CID 7528894.
- ^ д'Юдеваль С., Самнер С.Дж. (апрель 2015 г.). «Терапия спинальной мышечной атрофии: где мы находимся?». Нейротерапия. 12 (2): 303–16. Дои:10.1007 / s13311-015-0337-y. ЧВК 4404440. PMID 25631888.
- ^ «Генная терапия Novartis стоимостью 2,1 миллиона долларов должна стать самым дорогим лекарством в мире». Хранитель. Рейтер. 25 мая 2019. ISSN 0261-3077.
- ^ Benkhelifa-Ziyyat S, Besse A, Roda M, Duque S, Astord S, Carcenac R, Marais T., Barkats M (февраль 2013 г.). «Внутримышечная инъекция scAAV9-SMN опосредует широко распространенную доставку генов в спинной мозг и снижает тяжесть заболевания у мышей со SMA». Молекулярная терапия. 21 (2): 282–90. Дои:10.1038 / mt.2012.261. ЧВК 3594018. PMID 23295949.
- ^ "Biogen выпускает заявление сообщества о доступе к Spinraza и новых данных | Лечение SMA". www.curesma.org. Получено 11 сентября 2018.
- ^ «Новартис представляет обновленную информацию о клинических испытаниях LMI070 (Бранаплам)». CureSMA. Получено 7 октября 2017.
- ^ Клецл, Хайдемари; Марке, Энн; Гюнтер, Андреас; Тан, Вакана; Хойбергер, Жюль; Греневельд, Герт Ян; Биркгоф, Виллем; Меркури, Эухенио; Лохмюллер, Ханс; Вуд, Клэр; Фишер, Дирк; Герлах, Ирэн; Хайниг, Катя; Бугаван, Теодорика; Дзиадек, Себастьян; Кинч, Рассел; Чешский, христианский; Ходжа, Омар (2019). «Модификатор для орального сплайсинга RG7800 увеличивает выживаемость полной длины мРНК двигательного нейрона 2 и выживаемость белка двигательного нейрона: результаты испытаний на здоровых взрослых и пациентов со спинальной мышечной атрофией». Нервно-мышечные расстройства. Elsevier BV. 29 (1): 21–29. Дои:10.1016 / j.nmd.2018.10.001. ISSN 0960-8966. PMID 30553700. S2CID 54315649.
- ^ Чжан М.Л., Лорсон С.Л., Андрофи Э.Дж., Чжоу Дж. (Октябрь 2001 г.). «Репортерная система in vivo для измерения повышенного включения экзона 7 в мРНК SMN2: потенциальная терапия SMA». Генная терапия. 8 (20): 1532–8. Дои:10.1038 / sj.gt.3301550. PMID 11704813.
- ^ Андреасси К., Джареки Дж., Чжоу Дж., Куверт Д. Д., Монани У. Р., Чен Х, Уитни М., Поллок Б., Чжан М., Андрофи Е., Берджес А. Х. (ноябрь 2001 г.). «Лечение акларубицином восстанавливает уровни SMN в клетках, полученных от пациентов с мышечной атрофией I типа». Молекулярная генетика человека. 10 (24): 2841–9. Дои:10.1093 / hmg / 10.24.2841. PMID 11734549.
- ^ Чжоу Х, Мэн Дж, Марросу Э, Джангра Н, Морган Дж, Мунтони Ф (ноябрь 2015 г.). «Повторные низкие дозы морфолиноантисмыслового олигомера: промежуточная модель мышечной атрофии на мышах для изучения окна терапевтического ответа». Молекулярная генетика человека. 24 (22): 6265–77. Дои:10.1093 / hmg / ddv329. ЧВК 4614699. PMID 26264577.
- ^ Хаммонд С.М., Хейзелл Дж., Шабанпур Ф., Салех А.Ф., Бауэрман М., Сани Дж. Н., Мейбум К.Э., Чжоу Х., Мунтони Ф., Талбот К., Гейт М.Дж., Вуд М.Дж. (сентябрь 2016 г.). «Системная пептидопосредованная олигонуклеотидная терапия улучшает долгосрочную выживаемость при спинальной мышечной атрофии». Труды Национальной академии наук Соединенных Штатов Америки. 113 (39): 10962–7. Дои:10.1073 / pnas.1605731113. ЧВК 5047168. PMID 27621445.
- ^ Анджелоцци К., Борго Ф., Тициано Ф. Д., Мартелла А., Нери Дж., Браге С. (январь 2008 г.). «Сальбутамол увеличивает уровень мРНК SMN и белка в клетках с мышечной атрофией спинного мозга». Журнал медицинской генетики. 45 (1): 29–31. Дои:10.1136 / jmg.2007.051177. PMID 17932121. S2CID 29911453.
- ^ Пан M, Staccioli S, Messina S, D'Amico A, Pelliccioni M, Mazzone ES, Cuttini M, Alfieri P, Battini R, Main M, Muntoni F, Bertini E, Villanova M, Mercuri E (июль 2008 г.). «Ежедневный прием сальбутамола у молодых пациентов со СМА II типа». Нервно-мышечные расстройства. 18 (7): 536–40. Дои:10.1016 / j.nmd.2008.05.004. PMID 18579379. S2CID 34334434.
- ^ Тициано Ф. Д., Ломастро Р., Пинто А. М., Мессина С., Д'Амико А., Фиори С., Анджелоцци С., Пейн М, Меркури Е., Бертини Е., Нери Дж., Браге С. (декабрь 2010 г.). «Сальбутамол увеличивает уровни транскриптов мотонейронов выживаемости (SMN) в лейкоцитах пациентов с спинальной мышечной атрофией (СМА): актуальность для дизайна клинических испытаний» (PDF). Журнал медицинской генетики. 47 (12): 856–8. Дои:10.1136 / jmg.2010.080366. PMID 20837492. S2CID 21825049.
- ^ Моранди Л., Абиуси Е., Пасаниси МБ, Ломастро Р., Фиори С., Ди Пьетро Л., Анджелини С., Сорару Г., Гайани А., Монджини Т., Верчелли Л. (2013). «P.6.4 Переносимость и эффективность сальбутамола у взрослых пациентов с СМА III типа: результаты многоцентрового молекулярного и клинического двойного слепого плацебо-контролируемого исследования». Нервно-мышечные расстройства. 23 (9–10): 771. Дои:10.1016 / j.nmd.2013.06.475. S2CID 54398218.
- ^ Чанг Дж. Г., Се-Ли Х. М., Йонг Й. Дж., Ванг Н. М., Цай СН, Ли Х. (август 2001 г.). «Лечение спинальной мышечной атрофии бутиратом натрия». Труды Национальной академии наук Соединенных Штатов Америки. 98 (17): 9808–13. Bibcode:2001PNAS ... 98.9808C. Дои:10.1073 / pnas.171105098. ЧВК 55534. PMID 11504946.
- ^ Андреасси С., Анджелоцци С., Тициано Ф. Д., Витали Т., Де Винченци Е., Бонинсенья А., Вилланова М., Бертини Е., Пини А., Нери Дж., Браге С. (январь 2004 г.). «Фенилбутират увеличивает экспрессию SMN in vitro: актуальность для лечения спинальной мышечной атрофии». Европейский журнал генетики человека. 12 (1): 59–65. Дои:10.1038 / sj.ejhg.5201102. PMID 14560316.
- ^ Браге С., Виталий Т., Тициано Ф.Д., Анджелоцци С., Пинто А.М., Борго Ф., Москато У., Бертини Э., Меркури Э., Нери Дж. (Февраль 2005 г.). «Фенилбутират увеличивает экспрессию гена SMN у пациентов с спинальной мышечной атрофией». Европейский журнал генетики человека. 13 (2): 256–9. Дои:10.1038 / sj.ejhg.5201320. PMID 15523494.
- ^ Меркури Э, Бертини Э, Мессина С, Солари А, Д'Амико А, Анджелоцци С., Баттини Р., Берардинелли А, Боффи П, Бруно С., Чини С, Колитто Ф, Кинали М, Минетти С, Монджини Т, Моранди Л., Нери Дж., Орчези С., Пейн М, Пелличчони М., Пини А., Тициано Ф. Д., Вилланова М., Вита Дж., Браге С. (январь 2007 г.). «Рандомизированное двойное слепое плацебо-контролируемое испытание фенилбутирата при спинальной мышечной атрофии». Неврология. 68 (1): 51–5. Дои:10.1212 / 01.wnl.0000249142.82285.d6. PMID 17082463. S2CID 30429093.
- ^ Номер клинического исследования NCT00528268 для «Исследования по оценке фенилбутирата натрия у младенцев с предсимптоматической формой спинальной мышечной атрофии (STOPSMA)» на ClinicalTrials.gov
- ^ Брихта Л., Хофманн Ю., Ханен Э., Зибзехнрубль Ф.А., Рашке Х., Блюмке И., Эйюпоглу И. Ю., Вирт Б. (октябрь 2003 г.). «Вальпроевая кислота увеличивает уровень белка SMN2: препарат, хорошо известный как потенциальное средство для лечения спинальной мышечной атрофии». Молекулярная генетика человека. 12 (19): 2481–9. Дои:10.1093 / hmg / ddg256. PMID 12915451.
- ^ Цай Л.К., Цай М.С., Тинг С.Х., Ли Х. (ноябрь 2008 г.). «Множественные терапевтические эффекты вальпроевой кислоты у мышей модели спинальной мышечной атрофии». Журнал молекулярной медицины. 86 (11): 1243–54. Дои:10.1007 / s00109-008-0388-1. PMID 18649067. S2CID 24565272.
- ^ Свобода KJ, Скотт CB, Кроуфорд TO, Simard LR, Reyna SP, Krosschell KJ, Acsadi G, Elsheik B, Schroth MK, D'Anjou G, LaSalle B, Prior TW, Sorenson SL, Maczulski JA, Bromberg MB, Chan GM, Kissel JT, et al. (Сеть исследователей по проекту «Лечение спинальной мышечной атрофии») (август 2010 г.). Бутрон I (ред.). «Исследование SMA CARNI-VAL, часть I: двойное слепое рандомизированное плацебо-контролируемое испытание L-карнитина и вальпроевой кислоты при спинальной мышечной атрофии». PLOS ONE. 5 (8): e12140. Bibcode:2010PLoSO ... 512140S. Дои:10.1371 / journal.pone.0012140. ЧВК 2924376. PMID 20808854.
- ^ Киссель Дж. Т., Скотт С. Б., Рейна С. П., Кроуфорд ТО, Симард Л. Р., Кроссчелл К. Дж., Асади Дж., Эльшейк Б., Шрот М.К., Д'Анжу Дж., Ласалле Б., Приор Т.В., Соренсон С., Макзульски Дж. Свобода К.Дж. и др. (Сеть исследователей по проекту «Лечение спинальной мышечной атрофии») (2011). «SMA CARNIVAL TRIAL, ЧАСТЬ II: проспективное однорукое испытание L-карнитина и вальпроевой кислоты у амбулаторных детей со спинальной мышечной атрофией». PLOS ONE. 6 (7): e21296. Bibcode:2011PLoSO ... 621296K. Дои:10.1371 / journal.pone.0021296. ЧВК 3130730. PMID 21754985.
- ^ Дарбар И.А., Плаггерт П.Г., Резенде МБ, Занотели Э., Рид, Калифорния (март 2011 г.). «Оценка силы мышц и двигательных способностей у детей с атрофией мышц позвоночника II и III типов, получавших вальпроевую кислоту». BMC Neurology. 11: 36. Дои:10.1186/1471-2377-11-36. ЧВК 3078847. PMID 21435220.
- ^ Гарбес Л., Хеесен Л., Хёлькер И., Бауэр Т., Шремл Дж., Циммерманн К., Тонес М., Вальтер М., Димос Дж., Пейтц М., Брюстле О., Хеллер Р., Вирт Б. (январь 2013 г.). «Ответ VPA при SMA подавляется транслоказой жирных кислот CD36». Молекулярная генетика человека. 22 (2): 398–407. Дои:10.1093 / hmg / dds437. PMID 23077215.
- ^ Рак К., Лехнер Б.Д., Шнайдер С., Дрексл Х., Сендтнер М., Яблонка С. (декабрь 2009 г.). «Вальпроевая кислота блокирует возбудимость в моторных нейронах мышей SMA I типа». Нейробиология болезней. 36 (3): 477–87. Дои:10.1016 / j.nbd.2009.08.014. PMID 19733665. S2CID 34657615.
- ^ Эльшафай А., Хиеу Т.Х., Дохейм М.Ф., Кассем М.А., Эльдоадоа М.Ф., Холлоуэй С.К., Або-Эльгар Х., Хираяма К., Хай Н.Т. (март 2019 г.). «Эффективность и безопасность вальпроевой кислоты при спинальной мышечной атрофии: систематический обзор и метаанализ». Препараты ЦНС. 33 (3): 239–250. Дои:10.1007 / s40263-019-00606-6. PMID 30796634. S2CID 73495750.
- ^ Гржещик С.М., Ганта М., Приор Т.В., Хевлин В.Д., Ван С.Х. (август 2005 г.). «Гидроксимочевина усиливает экспрессию гена SMN2 в клетках с мышечной атрофией спинного мозга». Анналы неврологии. 58 (2): 194–202. Дои:10.1002 / ana.20548. PMID 16049920.
- ^ Chen TH, Chang JG, Yang YH, Mai HH, Liang WC, Wu YC, Wang HY, Huang YB, Wu SM, Chen YC, Yang SN, Jong YJ (декабрь 2010 г.). «Рандомизированное двойное слепое плацебо-контролируемое испытание гидроксимочевины при спинальной мышечной атрофии». Неврология. 75 (24): 2190–7. Дои:10.1212 / WNL.0b013e3182020332. PMID 21172842. S2CID 25858890.
- ^ Эванс М.С., Черри Дж. Дж., Андрофи Е. Дж. (Октябрь 2011 г.). «Дифференциальная регуляция гена SMN2 отдельными белками HDAC». Сообщения о биохимических и биофизических исследованиях. 414 (1): 25–30. Дои:10.1016 / j.bbrc.2011.09.011. ЧВК 6538936. PMID 21925145.
- ^ Riessland M, Brichta L, Hahnen E, Wirth B (август 2006 г.). «Бензамид M344, новый ингибитор гистондеацетилазы, значительно увеличивает уровни РНК / белка SMN2 в клетках с мышечной атрофией спинного мозга». Генетика человека. 120 (1): 101–10. Дои:10.1007 / s00439-006-0186-1. PMID 16724231. S2CID 24804136.
- ^ Гарбес Л., Риссланд М., Хёлькер И., Хеллер Р., Хауке Дж., Транкле С., Корас Р., Блюмке И., Ханен Е., Вирт Б. (октябрь 2009 г.). «LBH589 индуцирует до 10-кратного уровня белка SMN с помощью нескольких независимых механизмов и эффективен даже в клетках пациентов с СМА, не реагирующих на вальпроат». Молекулярная генетика человека. 18 (19): 3645–58. Дои:10.1093 / hmg / ddp313. PMID 19584083.
- ^ Narver HL, Kong L, Burnett BG, Choe DW, Bosch-Marcé M, Taye AA, Eckhaus MA, Sumner CJ (октябрь 2008 г.). «Устойчивое улучшение спинальной мышечной атрофии у мышей, получавших трихостатин А плюс питание». Анналы неврологии. 64 (4): 465–70. Дои:10.1002 / ana.21449. PMID 18661558.
- ^ Авила А.М., Бернетт Б.Г., Тайе А.А., Габанелла Ф., Найт М.А., Хартенштейн П., Джизман З., Ди Просперо Н.А., Пеллиццони Л., Фишбек К.Х., Самнер С.Дж. (март 2007 г.). «Трихостатин А увеличивает экспрессию SMN и выживаемость в мышиной модели спинальной мышечной атрофии». Журнал клинических исследований. 117 (3): 659–71. Дои:10.1172 / JCI29562. ЧВК 1797603. PMID 17318264.
- ^ Riessland M, Ackermann B, Förster A, Jakubik M, Hauke J, Garbes L, Fritzsche I, Mende Y, Blumcke I, Hahnen E, Wirth B (апрель 2010 г.). «SAHA улучшает фенотип SMA в двух моделях спинальной мышечной атрофии на мышах». Молекулярная генетика человека. 19 (8): 1492–506. Дои:10.1093 / hmg / ddq023. PMID 20097677.
- ^ Фарук Ф., Молина Ф.А., Хэдвен Дж., Маккензи Д., Уизерспун Л., Осмонд М., Холчик М., Маккензи А. (август 2011 г.). «Пролактин увеличивает экспрессию SMN и увеличивает выживаемость в мышиной модели тяжелой спинальной мышечной атрофии через путь STAT5». Журнал клинических исследований. 121 (8): 3042–50. Дои:10.1172 / JCI46276. ЧВК 3148738. PMID 21785216.
- ^ Сакла М.С., Лорсон С.Л. (январь 2008 г.). "Индукция полноразмерного выживания мотонейрона полифенольными растительными соединениями". Генетика человека. 122 (6): 635–43. Дои:10.1007 / s00439-007-0441-0. PMID 17962980. S2CID 12460406.
- ^ Даянгач-Эрден Д., Бора Дж., Айхан П., Коджафе С., Далкара С., Елекчи К., Демир А.С., Эрдем-Юртер Х. (март 2009 г.). «Активность ингибирования гистоновой деацетилазы и молекулярная стыковка (е) -ресвератрола: его терапевтический потенциал при спинальной мышечной атрофии». Химическая биология и дизайн лекарств. 73 (3): 355–64. CiteSeerX 10.1.1.515.8424. Дои:10.1111 / j.1747-0285.2009.00781.x. PMID 19207472.
- ^ Фарук Ф., Абадия-Молина Ф., Маккензи Д., Хадвен Дж., Шамим Ф., О'Рейли С., Хольчик М., Маккензи А. (сентябрь 2013 г.). «Целекоксиб увеличивает SMN и выживаемость в мышиной модели с тяжелой спинальной мышечной атрофией посредством активации пути p38». Молекулярная генетика человека. 22 (17): 3415–24. Дои:10.1093 / hmg / ddt191. PMID 23656793.
- ^ Бернетт Б.Г., Муньос Э., Тандон А., Квон Д.Й., Самнер С.Дж., Фишбек К.Х. (март 2009 г.). «Регулирование стабильности белка SMN». Молекулярная и клеточная биология. 29 (5): 1107–15. Дои:10.1128 / MCB.01262-08. ЧВК 2643817. PMID 19103745.
- ^ Мэттис В.Б., Рай Р., Ван Дж., Чанг К.В., Коади Т., Лорсон С.Л. (ноябрь 2006 г.). «Новые аминогликозиды повышают уровень SMN в фибробластах с мышечной атрофией спинного мозга». Генетика человека. 120 (4): 589–601. Дои:10.1007 / s00439-006-0245-7. PMID 16951947. S2CID 28834037.
- ^ Мэттис В.Б., Фоссо М.Ю., Чанг С.В., Лорсон С.Л. (ноябрь 2009 г.). «Подкожное введение TC007 снижает тяжесть заболевания на животной модели SMA». BMC Neuroscience. 10: 142. Дои:10.1186/1471-2202-10-142. ЧВК 2789732. PMID 19948047.
- ^ Lunn MR, Root DE, Martino AM, Flaherty SP, Kelley BP, Coovert DD, Burghes AH, Man NT, Morris GE, Zhou J, Androphy EJ, Sumner CJ, Stockwell BR (ноябрь 2004 г.). «Индопрофен активизирует белок выживания моторных нейронов посредством независимого от циклооксигеназы механизма». Химия и биология. 11 (11): 1489–93. Дои:10.1016 / j.chembiol.2004.08.024. ЧВК 3160629. PMID 15555999.
- ^ Тейлор Н.П. (1 июня 2018 г.). "Рош отменяет лекарство от СМА стоимостью 120 млн евро после многих трудностей"'". www.fiercebiotech.com. Получено 8 июн 2018.
- ^ Такеучи Y, Мияномае Y, Komatsu H, Oomizono Y, Nishimura A, Okano S, Nishiki T., Sawada T. (июль 1994 г.). «Эффективность тиреотропин-рилизинг-гормона в лечении мышечной атрофии позвоночника». Журнал детской неврологии. 9 (3): 287–9. Дои:10.1177/088307389400900313. PMID 7930408. S2CID 41678161.
- ^ Ценг А.С., Ченг Дж., Фричински Х., Ниранджан В., Ститик Т., Сиал А, Такеучи Й., Фойе П., ДеПринс М., Бах-младший (2000). «Исследование тиреотропин-рилизинг гормона для лечения спинальной мышечной атрофии: предварительный отчет». Американский журнал физической медицины и реабилитации. 79 (5): 435–40. Дои:10.1097/00002060-200009000-00005. PMID 10994885. S2CID 20416253.
- ^ Като З., Окуда М., Окумура Ю., Араи Т., Терамото Т., Нисимура М., Канеко Н., Кондо Н. (август 2009 г.). «Пероральное введение аналога тиреотропин-рилизинг гормона (TRH), талтирелина гидрата, при спинальной мышечной атрофии». Журнал детской неврологии. 24 (8): 1010–2. Дои:10.1177/0883073809333535. PMID 19666885. S2CID 29321906.
- ^ Wadman RI, Bosboom WM, van den Berg LH, Wokke LH, Iannaccone ST, Vrancken AF, et al. (Кокрановское сотрудничество) (7 декабря 2011 г.). Вадман Р.И. (ред.). «Медикаментозное лечение спинальной мышечной атрофии I типа». Кокрановская база данных систематических обзоров. John Wiley & Sons, Ltd (12): CD006281. Дои:10.1002 / 14651858.cd006281.pub3. PMID 22161399.
- ^ Хаддад Х., Сифуэнтес-Диас С., Мироглио А., Роблот Н., Джоши В., Мелки Дж. (Октябрь 2003 г.). «Рилузол ослабляет прогрессирование мышечной атрофии позвоночника на мышиной модели». Мышцы и нервы. 28 (4): 432–7. Дои:10.1002 / mus.10455. PMID 14506714.
- ^ Dimitriadi M, Kye MJ, Kalloo G, Yersak JM, Sahin M, Hart AC (апрель 2013 г.). «Нейропротекторное лекарственное средство рилузол действует через Са2 + -активированные каналы K + с небольшой проводимостью, улучшая дефекты в моделях спинальной мышечной атрофии».. Журнал неврологии. 33 (15): 6557–62. Дои:10.1523 / JNEUROSCI.1536-12.2013. ЧВК 3652322. PMID 23575853.
- ^ Номер клинического исследования NCT00774423 за «Исследование по оценке эффективности рилузола у детей и подростков со спинальной мышечной атрофией (СМА)» в ClinicalTrials.gov
- ^ "Riluzole: décevants premiers résultats" (На французском). AFM Téléthon. 22 сентября 2010 г.
- ^ Низардо М., Нардини М., Рончи Д., Салани С., Донадони С., Фортунато Ф, Колчиаго Дж., Фальконе М., Симоне С., Рибольди Дж., Говони А., Брезолин Н., Коми ГП, Корти С. (июнь 2011 г.). «Бета-лактамный антибиотик предлагает нейрозащиту в модели спинальной мышечной атрофии за счет нескольких механизмов» (PDF). Экспериментальная неврология. 229 (2): 214–25. Дои:10.1016 / j.expneurol.2011.01.017. HDL:2434/425410. PMID 21295027. S2CID 47567316.
- ^ Хедлунд Э (сентябрь 2011 г.). «Защитные эффекты β-лактамных антибиотиков при нарушениях двигательных нейронов». Экспериментальная неврология. 231 (1): 14–8. Дои:10.1016 / j.expneurol.2011.06.002. PMID 21693120. S2CID 26353910.
- ^ Роуз Ф.Ф., Мэттис В.Б., Риндт Х., Лорсон К.Л. (март 2009 г.). «Доставка рекомбинантного фоллистатина снижает тяжесть заболевания на мышиной модели спинальной мышечной атрофии». Молекулярная генетика человека. 18 (6): 997–1005. Дои:10.1093 / hmg / ddn426. ЧВК 2649020. PMID 19074460.
- ^ «СК-2127107».
- ^ Карроцци М., Амаддео А., Бионди А., Занус С., Монти Ф., Алессандро В. (ноябрь 2012 г.). «Стволовые клетки при тяжелой детской спинномозговой мышечной атрофии (SMA1)». Нервно-мышечные расстройства. 22 (11): 1032–4. Дои:10.1016 / j.nmd.2012.09.005. PMID 23046997. S2CID 42093152.
- ^ Меркури Э., Бертини Э. (декабрь 2012 г.). «Стволовые клетки при тяжелой детской спинальной мышечной атрофии». Нервно-мышечные расстройства. 22 (12): 1105. Дои:10.1016 / j.nmd.2012.11.001. PMID 23206850. S2CID 43858783.
- ^ Комитет по передовой терапии и Научный секретариат CAT (август 2010 г.). «Использование нерегулируемых лекарственных средств на основе стволовых клеток». Ланцет. 376 (9740): 514. Дои:10.1016 / S0140-6736 (10) 61249-4. PMID 20709228. S2CID 6906599.
- ^ Европейское агентство по лекарствам (16 апреля 2010 г.). «Обеспокоенность по поводу нерегулируемых лекарственных средств, содержащих стволовые клетки» (PDF). Европейское агентство по лекарствам.
- ^ «Национальные реестры DMD, SMA и DM». Архивировано из оригинал 22 января 2011 г.
дальнейшее чтение
- Парано Е., Павоне Л., Фальсаперла Р., Трифилетти Р., Ван С. (август 1996 г.). «Молекулярные основы фенотипической гетерогенности братьев и сестер со спинальной мышечной атрофией». Анналы неврологии. 40 (2): 247–51. Дои:10.1002 / ana.410400219. PMID 8773609.
- Ван Ч., Финкель Р.С., Бертини Э.С., Шрот М., Саймондс А., Вонг Б., Алоизиус А., Моррисон Л., Мейн М., Кроуфорд ТО, Трела А. (август 2007 г.). «Заявление о консенсусе по стандарту лечения спинальной мышечной атрофии». Журнал детской неврологии. 22 (8): 1027–49. Дои:10.1177/0883073807305788. PMID 17761659. S2CID 6478040.
внешняя ссылка
| Классификация | |
|---|---|
| Внешние ресурсы |