Рибозо-фосфатдифосфокиназа - Ribose-phosphate diphosphokinase
| Рибозо-фосфатдифосфокиназа | |||||||||
|---|---|---|---|---|---|---|---|---|---|
 Фосфорибозилпирофосфатсинтаза 1, гексамер, человек | |||||||||
| Идентификаторы | |||||||||
| Номер ЕС | 2.7.6.1 | ||||||||
| Количество CAS | 9031-46-3 | ||||||||
| Базы данных | |||||||||
| IntEnz | Просмотр IntEnz | ||||||||
| БРЕНДА | BRENDA запись | ||||||||
| ExPASy | Просмотр NiceZyme | ||||||||
| КЕГГ | Запись в KEGG | ||||||||
| MetaCyc | метаболический путь | ||||||||
| ПРИАМ | профиль | ||||||||
| PDB структуры | RCSB PDB PDBe PDBsum | ||||||||
| Генная онтология | AmiGO / QuickGO | ||||||||
| |||||||||
| фосфорибозилпирофосфат синтетаза 1 | |||||||
|---|---|---|---|---|---|---|---|
| Идентификаторы | |||||||
| Символ | PRPS1 | ||||||
| Ген NCBI | 5631 | ||||||
| HGNC | 9462 | ||||||
| OMIM | 311850 | ||||||
| RefSeq | NM_002764 | ||||||
| UniProt | P60891 | ||||||
| Прочие данные | |||||||
| Номер ЕС | 2.7.6.1 | ||||||
| Locus | Chr. Икс q21-q27 | ||||||
| |||||||
| фосфорибозилпирофосфат синтетаза 2 | |||||||
|---|---|---|---|---|---|---|---|
| Идентификаторы | |||||||
| Символ | PRPS2 | ||||||
| Ген NCBI | 5634 | ||||||
| HGNC | 9465 | ||||||
| OMIM | 311860 | ||||||
| RefSeq | NM_002765 | ||||||
| UniProt | P11908 | ||||||
| Прочие данные | |||||||
| Номер ЕС | 2.7.6.1 | ||||||
| Locus | Chr. Икс pter-q21 | ||||||
| |||||||
Рибозо-фосфатдифосфокиназа (или же фосфорибозилпирофосфатсинтетаза или же рибозо-фосфатпирофосфокиназа) является фермент что обращает рибозо-5-фосфат в фосфорибозилпирофосфат (ПРПП).[1][2] Классифицируется под EC 2.7.6.1.
Фермент участвует в синтезе нуклеотиды (пурины и пиримидины ), кофакторы НАД и НАДФ, и аминокислоты гистидин и триптофан,[1][2][3] связывание этих биосинтетических процессов с пентозофосфатным путем, из которого происходит субстрат рибозо-5-фосфат. Рибозо-5-фосфат производится Шунтирующий тракт HMP из Глюкозо-6-фосфат. Продукт фосфорибозилпирофосфат действует как важный компонент Пуриновый путь спасения и синтез пуринов de novo. Таким образом, дисфункция фермента подорвет метаболизм пуринов. Рибозофосфатпирофосфокиназа существует в бактериях, растениях и животных, и существует три изоформы человеческой рибозофосфатпирофосфокиназы.[2] У человека гены, кодирующие фермент, расположены на Х хромосома.[2]
Механизм реакции

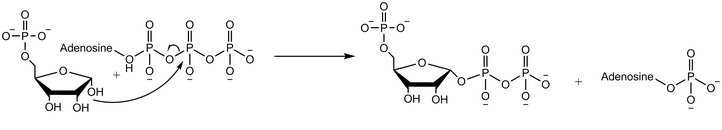
Рибозо-фосфатдифосфокиназа переносит дифосфорильную группу с Mg-ATP (Mg2 +, координированный с ATP) на рибозо-5-фосфат.[2] Ферментативная реакция начинается со связывания рибозо-5-фосфата с последующим связыванием Mg-ATP с ферментом. В переходном состоянии при связывании обоих субстратов происходит перенос дифосфата. Фермент сначала высвобождает АМФ, прежде чем высвободить фосфорибозилпирофосфат.[4]Эксперименты с использованием воды, меченной кислородом 18, демонстрируют, что механизм реакции протекает с нуклеофильной атакой аномерной гидроксильной группы рибозо-5-фосфата на бета-фосфор АТФ в SN2 реакция.[5]
Структура

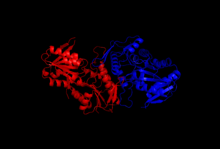
Кристаллизация и исследования дифракции рентгеновских лучей прояснили структуру фермента, который был выделен методами клонирования, экспрессии белка и очистки. Одна субъединица рибозо-фосфатдифосфокиназы состоит из 318 аминокислот; Активный ферментный комплекс состоит из трех гомодимеров (или шести субъединиц, гексамера). Структура одной субъединицы представляет собой пятицепочечную параллельную бета-лист (центральное ядро) в окружении четырех альфа спирали на N-концевой домен и пять альфа-спиралей на С-концевой домен, с двумя короткими антипараллельными бета-листами, отходящими от ядра.[2]Каталитический сайт фермента связывает АТФ и рибозо-5-фосфат. Гибкая петля (Phe92-Ser108), пирофосфат-связывающая петля (Asp171-Gly174) и флаговая область (Val30-Ile44 от соседней субъединицы) составляют сайт связывания АТФ, расположенный на границе раздела между двумя доменами одной субъединицы. Гибкая петля названа так из-за большой изменчивости строения.[6] Сайт связывания рибозо-5-фосфата состоит из остатков Asp220 – Thr228, расположенных в С-концевом домене одной субъединицы.[2][6]Аллостерический сайт, связывающий АДФ, состоит из аминокислотных остатков из трех субъединиц.[2]
Функция
Продукт этой реакции, фосфорибозилпирофосфат (PRPP), используется во многих биосинтезе (de novo и спасение ) пути. PRPP обеспечивает синтез рибозы de novo пуринов и пиримидинов, используемых в нуклеотидных основаниях, которые образуют РНК и ДНК. PRPP реагирует с ругать с образованием оротидилата, который может быть преобразован в уридилат (UMP). Затем UMP можно превратить в нуклеотид цитидинтрифосфат (ОСАГО). Реакция PRPP, глутамина и аммиака приводит к образованию 5-фосфорибозил-1-амина, предшественника инозинат (IMP), который в конечном итоге может быть преобразован в аденозинтрифосфат (ATP) или гуанозинтрифосфат (GTP). PRPP играет роль в путях спасения пуринов, реагируя со свободными пуриновыми основаниями с образованием аденилата, гуанилата и инозината.[7][8] PRPP также используется в синтезе НАД: реакция PRPP с никотиновой кислотой дает промежуточный мононуклеотид никотиновой кислоты.[9]
Регулирование
Рибозо-фосфатдифосфокиназа требует Mg2 + для активности; фермент действует только на АТФ согласовано с Mg2 +. Рибозо-фосфатдифосфокиназа регулируется фосфорилированием и аллостерией. Он активируется фосфат и тормозится ADP; предполагается, что фосфат и АДФ конкурируют за один и тот же регуляторный сайт. При нормальной концентрации фосфат активирует фермент, связываясь с его аллостерическим регуляторным сайтом. Однако показано, что при высоких концентрациях фосфат оказывает ингибирующее действие, конкурируя с субстратом рибозо-5-фосфатом за связывание в активном центре. АДФ является ключевым аллостерическим ингибитором рибозо-фосфатдифосфокиназы. Было показано, что при более низких концентрациях субстрата рибозо-5-фосфата АДФ может конкурентно ингибировать фермент. Рибозо-фосфатпирофосфокиназа также ингибируется некоторыми из продуктов ее биосинтеза.[2][6]
Роль в болезни
Поскольку ее продукт является ключевым соединением во многих путях биосинтеза, рибозо-фосфатдифосфокиназа участвует в некоторых редких заболеваниях и Х-сцепленные рецессивные заболевания. Мутации, которые приводят к сверхактивности (повышение активности фермента или нарушение регуляции фермента), приводят к перепроизводству пурина и мочевой кислоты. Симптомы сверхактивности включают: подагра, нейросенсорная тугоухость,[10] слабый мышечный тонус (гипотония), нарушение координации мышц (атаксия), наследственная периферическая нейропатия,[11] и расстройство нервного развития.[12][13][14] Мутации, которые приводят к потере функции рибозо-фосфатдифосфокиназы, приводят к Болезнь Шарко-Мари-Тута и АРТС-синдром.[15]
Рекомендации
- ^ а б Visentin LP, Hasnain S, Gallin W (июль 1977 г.). «Рибосомный белок S1 / S1A в бактериях». FEBS Lett. 79 (2): 258–63. Дои:10.1016/0014-5793(77)80799-0. PMID 330231.
- ^ а б c d е ж грамм час я Ли С., Лу И, Пэн Б., Дин Дж. (Январь 2007 г.). «Кристаллическая структура фосфорибозилпирофосфатсинтетазы 1 человека обнаруживает новый аллостерический сайт». Biochem. J. 401 (1): 39–47. Дои:10.1042 / BJ20061066. ЧВК 1698673. PMID 16939420.
- ^ Тан В., Ли Х, Чжу З., Тонг С., Ли Х, Чжан Х, Тэн М., Ню Л. (май 2006 г.). «Экспрессия, очистка, кристаллизация и предварительный рентгеноструктурный анализ человеческой фосфорибозилпирофосфат синтетазы 1 (PRS1)». Acta Crystallographica Раздел F. 62 (Пт 5): 432–4. Дои:10.1107 / S1744309106009067. ЧВК 2219982. PMID 16682768.
- ^ Фокс И. Х., Келли В. Н. (апрель 1972 г.). «Человеческая фосфорибозилпирофосфатсинтетаза. Кинетический механизм и ингибирование конечного продукта». J. Biol. Chem. 247 (7): 2126–31. PMID 4335863.
- ^ Миллер Г.А., Розенцвейг С., Свитцер Р.Л. (декабрь 1975 г.). «Кислород-18 исследования механизма переноса пирофосфорильной группы, катализируемого фосфорибозилпирофосфатсинтетазой». Arch. Biochem. Биофизы. 171 (2): 732–6. Дои:10.1016/0003-9861(75)90086-7. PMID 173242.
- ^ а б c Эриксен Т.А., Кадзиола А., Бентсен А.К., Харлоу К.В., Ларсен С. (апрель 2000 г.). «Структурные основы функции фосфорибозилпирофосфатсинтетазы Bacillus subtilis». Nat. Struct. Биол. 7 (4): 303–8. Дои:10.1038/74069. PMID 10742175.
- ^ Фокс И. Х., Келли В. Н. (март 1971 г.). «Фосфорибозилпирофосфат в человеке: биохимическое и клиническое значение». Анна. Междунар. Med. 74 (3): 424–33. Дои:10.7326/0003-4819-74-3-424. PMID 4324023.
- ^ Джереми М. Берг; Джон Л. Тимочко; Люберт Страйер; Грегори Дж. Гатто младший (2012). Биохимия (7-е изд.). Нью-Йорк: W.H. Фримен. ISBN 1429229365.
- ^ Ронгво А., Андрис Ф, Ван Гул Ф, Лео О (июль 2003 г.). «Реконструкция метаболизма НАД эукариот». BioEssays. 25 (7): 683–90. Дои:10.1002 / bies.10297. PMID 12815723.
- ^ Лю Х, Хан Д, Ли Дж, Хан Би, Оуян Х, Ченг Дж, Ли Х, Джин З, Ван Й, Битнер-Глинджич М., Конг Х, Сю Х, Кантарджиева А., Иви Р. Д., Сейдман К. Э., Сейдман Дж. Г., Du LL, Chen ZY, Dai P, Teng M, Yan D, Yuan H (январь 2010 г.). «Мутации с потерей функции в гене PRPS1 вызывают тип несиндромной Х-сцепленной нейросенсорной глухоты, DFN2». Являюсь. J. Hum. Genet. 86 (1): 65–71. Дои:10.1016 / j.ajhg.2009.11.015. ЧВК 2801751. PMID 20021999.
- ^ Kim HJ, Sohn KM, Shy ME, Krajewski KM, Hwang M, Park JH, Jang SY, Won HH, Choi BO, Hong SH, Kim BJ, Suh YL, Ki CS, Lee SY, Kim SH, Kim JW (сентябрь 2007 г. ). «Мутации в PRPS1, который кодирует фермент фосфорибозилпирофосфатсинтетазу, критический для биосинтеза нуклеотидов, вызывают наследственную периферическую невропатию с потерей слуха и оптической нейропатией (cmtx5)». Являюсь. J. Hum. Genet. 81 (3): 552–8. Дои:10.1086/519529. ЧВК 1950833. PMID 17701900.
- ^ Беккер М.А., Смит П.Р., Тейлор В., Мустафи Р., Свитцер Р.Л. (ноябрь 1995 г.). «Генетическая и функциональная основа повышенной активности фосфорибозилпирофосфатсинтетазы, устойчивой к пуриновым нуклеотидам». J. Clin. Вкладывать деньги. 96 (5): 2133–41. Дои:10.1172 / JCI118267. ЧВК 185862. PMID 7593598.
- ^ Зореф Э., Де Вриз А., Сперлинг О. (ноябрь 1975 г.). «Мутантная устойчивая к обратной связи фосфорибозилпирофосфатсинтетаза, связанная с избыточной продукцией пуринов и подагрой. Фосфорибозилпирофосфат и метаболизм пурина в культивируемых фибробластах». J. Clin. Вкладывать деньги. 56 (5): 1093–9. Дои:10.1172 / JCI108183. ЧВК 301970. PMID 171280.
- ^ «Повышенная активность фосфорибозилпирофосфатсинтетазы». Листер Хилл Национальный центр биомедицинских коммуникаций. Получено 25 февраля 2014.
- ^ Synofzik M, Müller Vom Hagen J, Haack TB, Wilhelm C, Lindig T., Beck-Wödl S, Nabuurs SB, van Kuilenburg AB, de Brouwer AP, Schöls L (2014). «Х-сцепленная болезнь Шарко-Мари-Тута, синдром Арса и предъязыковая несиндромальная глухота образуют континуум болезни: данные из семьи с новой мутацией PRPS1». Орфанет J Редкие Диск. 9 (1): 24. Дои:10.1186/1750-1172-9-24. ЧВК 3931488. PMID 24528855.
внешняя ссылка
- Uniprot - Рибозо-фосфатпирофосфокиназа 1
- GeneReviews / NIH / NCBI / UW запись о невропатии Шарко-Мари-Тута X типа 5
- Записи OMIM о невропатии Шарко-Мари-Тута X типа 5
- GeneReviews / NCBI / NIH / UW запись о синдроме искусств
- GeneReviews / NIH / NCBI / UW запись о суперактивности фосфорибозилпирофосфатсинтетазы (PRS)
- GeneReviews / NCBI / NIH / UW запись о несиндромной потере слуха и глухоте DFNX1
- Фосфорибозил + пирофосфат + синтетаза в Национальной медицинской библиотеке США Рубрики медицинской тематики (MeSH)