Болезнь Вильсона - Wilsons disease - Wikipedia
| Болезнь Вильсона | |
|---|---|
| Другие имена | Болезнь Вильсона, гепатолентикулярная дегенерация |
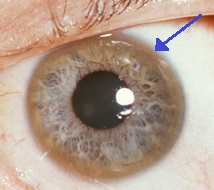 | |
| Коричневое кольцо на краю роговицы (Кольцо Кайзера – Флейшера ) часто встречается при болезни Вильсона, особенно при наличии неврологических симптомов. | |
| Специальность | Гастроэнтерология |
| Симптомы | Отек ног, желтоватая кожа, изменения личности[1] |
| Обычное начало | Возраст от 5 до 35 лет[1] |
| Причины | Генетический |
| Дифференциальная диагностика | Хроническая болезнь печени, болезнь Паркинсона, рассеянный склероз, другие[2][3] |
| Уход | Диетические изменения, хелатирующие агенты, добавки цинка, пересадка печени[1] |
| Частота | ~ 1 на 30 000[1] |
Болезнь Вильсона это генетическое расстройство в каком избытке медь накапливается в теле. Симптомы обычно связаны с мозг и печень. Симптомы, связанные с печенью, включают: рвота, слабое место, скопление жидкости в брюшной полости, отек ног, желтоватая кожа и зуд. Симптомы, связанные с мозгом, включают: тремор, ригидность мышц, проблемы с речью, изменения личности, беспокойство и психоз.[1]
Болезнь Вильсона вызывается мутация в Белок болезни Вильсона (ATP7B) ген. Этот белок транспортирует избыток меди в желчь, где он выводится с продуктами жизнедеятельности. Состояние аутосомно-рецессивный; Чтобы человек был поражен, он должен унаследовать мутированную копию гена от обоих родителей. Диагностика может быть сложной и часто включает в себя сочетание анализов крови, мочи и биопсия печени. Генетическое тестирование может использоваться для проверки членов семей пострадавших.[1]
Болезнь Вильсона обычно лечится с помощью диетических изменений и лекарств. Диетические изменения включают в себя диету с низким содержанием меди и отказ от медной посуды. Используемые лекарства включают: хелатирующие агенты Такие как триентин и d-пеницилламин и добавки цинка. Осложнения болезни Вильсона могут включать: отказ печени, рак печени и проблемы с почками. А пересадка печени может быть полезен тем, у кого другие методы лечения неэффективны, или при печеночной недостаточности.[1]
Болезнь Вильсона встречается примерно у 1 из 30 000 человек.[1] Симптомы обычно появляются в возрасте от 5 до 35 лет.[1] Впервые он был описан в 1854 году немецким патологом. Фридрих Теодор фон Фрерихс и назван в честь британского невролога Сэмюэл Уилсон.[4]
Признаки и симптомы
Основными очагами накопления меди являются печень и мозг, и, следовательно, заболевание печени и нейропсихиатрические симптомы являются основными признаками, которые приводят к постановке диагноза.[5] Люди с проблемами печени, как правило, обращаются за медицинской помощью раньше, как правило, в детстве или подростковом возрасте, чем люди с неврологическими и психиатрическими симптомами, которым обычно от двадцати лет и старше. Некоторые из них идентифицированы только потому, что у родственников была диагностирована болезнь Вильсона; у многих из них при тестировании выясняется, что у них наблюдаются симптомы этого состояния, но диагноз так и не был поставлен.[6]
Болезнь печени
Заболевание печени может проявляться как усталость, повышенная склонность к кровотечениям или спутанность сознания (из-за печеночная энцефалопатия ) и портальная гипертензия. Последнее, состояние, при котором давление в воротная вена заметно увеличивается, приводит к варикоз пищевода, кровеносные сосуды в пищевод которые могут кровоточить опасным для жизни образом, а также увеличивают селезенку (спленомегалия ) и скопление жидкости в брюшной полости (асцит ). При осмотре признаки хронического заболевания печени, такие как: ангиомы паука (небольшие расширенные кровеносные сосуды, обычно на груди). Хронический активный гепатит вызвало цирроз печени в большинстве случаев к тому времени, когда у них развиваются симптомы. Хотя у большинства людей с циррозом есть повышенный риск гепатоцеллюлярная карцинома (рак печени), этот риск относительно очень низок при болезни Вильсона.[5]
Около 5% всех людей диагностируются только тогда, когда у них развивается молниеносная острая печеночная недостаточность, часто в контексте гемолитическая анемия (анемия из-за разрушения эритроцитов). Это приводит к нарушениям в производстве белка (обнаруживаемым по нарушенным коагуляция ) и метаболизм печенью. Нарушение метаболизма белков приводит к накоплению продуктов жизнедеятельности, таких как аммиак в кровотоке. Когда это раздражает мозг, человек развивается печеночная энцефалопатия (спутанность сознания, кома, судороги и, наконец, опасные для жизни отек мозга ).[5]
Психоневрологические симптомы
Около половины людей с болезнью Вильсона имеют неврологические или психиатрические симптомы. У большинства изначально наблюдается легкое ухудшение когнитивных функций и неуклюжесть, а также изменения в поведении. Затем обычно следуют специфические неврологические симптомы, часто в виде паркинсонизм (жесткость зубчатого колеса, брадикинезия или замедленные движения и отсутствие равновесия - наиболее распространенные симптомы паркинсонизма[7]) с типичной рукой или без нее тремор, замаскированная мимика, невнятная речь, атаксия (отсутствие координации) или дистония (скручивания и повторяющиеся движения частью тела). Судороги и мигрень по-видимому, чаще встречается при болезни Вильсона.[5] Характерный тремор, описываемый как «тремор взмахов крыльев», встречается у многих людей с синдромом Вильсона; в покое этого нет, но можно спровоцировать отведением рук и сгибанием локтей к средней линии.[8]
Познание также может быть нарушено при болезни Вильсона. Он делится на две, не исключающие друг друга, категории: заболевание лобной доли (может быть представлен как импульсивность, нарушение суждения, распущенность, апатия и исполнительная дисфункция с плохим планированием и принятием решений) и подкорковая деменция (может проявляться замедленным мышлением, потерей памяти и исполнительная дисфункция, без признаков афазия, апраксия или же агнозия ). Предполагается, что эти когнитивные нарушения связаны и тесно связаны с психиатрическими проявлениями заболевания.[7]
Психиатрические проблемы из-за болезни Вильсона могут включать изменения в поведении, депрессия, тревожные расстройства, и психоз.[5] Психиатрические симптомы обычно сочетаются с неврологическими симптомами и редко проявляются сами по себе. Эти симптомы часто плохо определены и иногда могут быть связаны с другими причинами. Из-за этого диагноз болезни Вильсона редко ставится при наличии только психических симптомов.[7]
Другие системы органов
Заболевания связаны с накоплением меди при болезни Вильсона:
- Глаза: Кольца Кайзера – Флейшера (Кольца KF), a патогномоничный знак, может быть виден в роговица глаз, прямо или на щелевая лампа осмотр как отложения меди в кольце вокруг роговицы. Они связаны с отложением меди в Десцеметова мембрана. Эти кольца могут быть темно-коричневыми, золотистыми или красновато-зелеными, иметь ширину от 1 до 3 мм и появляться в области лимба роговицы. Они возникают не у всех людей с болезнью Вильсона. Болезнь Вильсона также связана с подсолнечником. катаракта проявляется коричневой или зеленой пигментацией передней и задней капсулы хрусталика.[9] Ни то, ни другое не вызывают значительной потери зрения.[5] Кольца KF встречаются примерно в 66% диагностированных случаев (чаще у пациентов с неврологическими симптомами, а не с проблемами печени).[6]
- Почки: почечный канальцевый ацидоз (Тип 2), расстройство бикарбонат обработка проксимальные канальцы приводит к нефрокальциноз (накопление кальция в почках), ослабление костей (из-за потери кальция и фосфатов), а иногда и аминоацидурия (потеря существенного аминокислоты необходим для синтеза белка).[5]
- Сердце: кардиомиопатия (слабость сердечной мышцы) - редкая, но признанная проблема при болезни Вильсона; это может привести к сердечная недостаточность (скопление жидкости из-за ухудшения работы насоса) и сердечные аритмии (эпизоды нерегулярного и / или ненормально быстрого или медленного сердцебиения).[5]
- Гормоны: гипопаратиреоз (отказ паращитовидные железы приводит к низкому уровню кальция), бесплодие, и повторный выкидыш.[5]

Катаракта подсолнечника и толстое кольцо KF у 40-летнего мужчины с болезнью Вильсона и декомпенсированной ХЗП

Рассеянное освещение роговицы

Отложение меди на десцеметовой мембране роговицы


Генетика

Ген болезни Вильсона (ATP7B) находится на хромосома 13 (13q14.3) и экспрессируется преимущественно в печени, почка, и плацента. Ген кодирует P-тип (фермент переноса катионов) АТФаза который транспортирует медь в желчь и включает его в церулоплазмин.[5] Мутации можно обнаружить в 90% случаев. Большинство (60%) гомозиготный за ATP7B мутации (две аномальные копии), и 30% имеют только одну аномальную копию. У десяти процентов нет обнаруживаемой мутации.[6]
Хотя 300 мутаций ATP7B были описаны, в большинстве популяций случаи болезни Вильсона возникают из-за небольшого количества мутаций, специфичных для этой популяции. Например, в западных популяциях мутация H1069Q (замена гистидин по глутамин в положении 1069 белка) присутствует в 37–63% случаев, в то время как в Китае эта мутация встречается очень редко и R778L (аргинин к лейцин 778) встречается чаще. Относительно мало известно об относительном влиянии различных мутаций, хотя, согласно некоторым исследованиям, мутация H1069Q, по-видимому, предсказывает более позднее начало и преимущественно неврологические проблемы.[5][10] Обширный ресурс с клиническими комментариями, WilsonGen предоставляет клиническую классификацию вариантов в соответствии с последними рекомендациями ACMG и AMP.[11]
Нормальная вариация в PRNP Ген может изменить течение болезни, отсрочив возраст начала и влияя на тип симптомов, которые развиваются. Этот ген производит прионный белок, который активен в головном мозге и других тканях, а также, по-видимому, участвует в транспортировке меди.[12] Роль для ApoE изначально предполагалось, но не удалось подтвердить.[10]
Состояние наследуется по аутосомно-рецессивному типу. Чтобы унаследовать его, оба родителя человека должны нести пораженный ген. Большинство из них не имеют семейного анамнеза этого заболевания.[10] Люди с одним аномальным геном называются носителями (гетерозиготами) и могут иметь легкие, но не значимые с медицинской точки зрения, нарушения метаболизма меди.[13]
Болезнь Вильсона является наиболее распространенной из группы наследственных заболеваний, вызывающих перегрузку медью в печени. Все может вызвать цирроз в молодости. Остальные члены группы Индийский цирроз в детстве (ICC), эндемический тирольский детский цирроз и идиопатический токсикоз меди. Это не связано с ATP7B мутации: например, ICC был связан с мутациями в KRT8 и KRT18 ген.[10]
Патофизиология
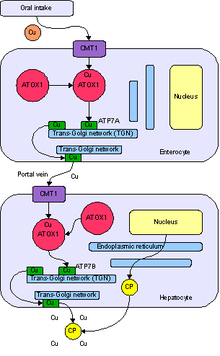
Медь нужна организму для количество функций, преимущественно как кофактор для ряда ферментов, таких как церулоплазмин, цитохром с оксидаза, дофамин-β-гидроксилаза, супероксиддисмутаза и тирозиназа.[10]
Медь попадает в организм через пищеварительный тракт. Белок-переносчик на клетки тонкой кишки, транспортер медной мембраны 1 (Ctr1; SLC31A1), переносит медь внутрь клеток, где некоторые из них связаны с металлотионеин и часть несет ATOX1 органелле, известной как сеть транс-Гольджи. Здесь, в ответ на повышение концентрации меди, фермент под названием ATP7A (Белок Менкеса) выделяет медь в воротная вена в печень. Клетки печени также несут белок CMT1, а металлотионеин и ATOX1 связывают его внутри клетки, но здесь именно ATP7B связывает медь с церулоплазмин и высвобождает ее в кровоток, а также удаляет излишки меди, выделяя ее в желчь. Обе функции ATP7B нарушены при болезни Вильсона. Медь накапливается в ткани печени; церулоплазмин все еще секретируется, но в форме, в которой отсутствует медь (так называемый апоцерулоплазмин), и быстро разлагается в кровотоке.[10]
Когда количество меди в печени превышает количество белков, которые обычно связывают ее, это вызывает окислительное повреждение в результате процесса, известного как Фентон химия; этот ущерб в конечном итоге приводит к хронический активный гепатит, фиброз (отложение соединительной ткани) и цирроз. Печень также выделяет в кровоток медь, которая не связана с церулоплазмином. Эта свободная медь осаждается по всему телу, но особенно в почках, глазах и мозге. В головном мозге большая часть меди откладывается в базальный ганглий, особенно в скорлупа и бледный шар (вместе называли чечевицеобразное ядро ); эти области обычно участвуют в координации движений, а также играют важную роль в нейрокогнитивных процессах, таких как обработка стимулов и регуляция настроения. Повреждение этих областей, опять-таки химическим методом Фентона, вызывает психоневрологические симптомы, наблюдаемые при болезни Вильсона.[10]
Неясно, почему болезнь Вильсона вызывает гемолиз, но различные данные свидетельствуют о том, что высокий уровень свободных (нецерулоплазмин связанная) медь оказывает прямое влияние на окисление гемоглобин, ингибирование энергоснабжающих ферментов в эритроцит, или прямое повреждение клеточная мембрана.[14]
Диагностика
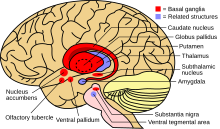
Болезнь Вильсона можно заподозрить на основании любого из симптомов, упомянутых выше, или если у близкого родственника обнаружена болезнь Вильсона. У большинства немного ненормальные функциональные пробы печени например поднятый аспартат трансаминаза, аланин трансаминаза и билирубин уровень. Если повреждение печени значительное, альбумин может снижаться из-за неспособности поврежденных клеток печени производить этот белок; аналогично, протромбиновое время (испытание коагуляция ) может продолжаться, так как печень неспособна вырабатывать белки, известные как факторы свертывания крови.[5] Щелочная фосфатаза уровни относительно низкие у пациентов с острой печеночной недостаточностью, связанной с Вильсоном.[15] Если есть неврологические симптомы, магнитно-резонансная томография (МРТ) головного мозга обычно выполняется; это показывает гиперинтенсивность в части мозга, называемой базальный ганглий в Т2 параметр.[13] МРТ также может продемонстрировать характерные "лицо гигантской панды" шаблон.[16]
Не существует полностью надежного теста на болезнь Вильсона, но уровни церулоплазмин и медь в крови, а также количество меди, выделяемой с мочой в течение 24-часового периода, вместе используются для формирования представления о количестве меди в организме. В Золотой стандарт - или самый идеальный тест - это биопсия печени.[5]
Церулоплазмин

Уровни церулоплазмин аномально низкие (<0,2 г / л) в 80–95% случаев.[5] Однако он может присутствовать на нормальном уровне у людей с постоянным воспаление как это белок острой фазы. Низкий уровень церулоплазмина также содержится в Болезнь Менкеса и ацерулоплазминемия, которые связаны с болезнью Вильсона, но гораздо реже.[5][13]
Сочетание неврологических симптомов, колец Кайзера-Флейшера и низкого уровня церулоплазмина считается достаточным для диагностики болезни Вильсона. Однако во многих случаях необходимы дальнейшие тесты.[13]
Медь в сыворотке и моче
Уровень меди в сыворотке крови низкий, что может показаться парадоксальным, учитывая, что болезнь Вильсона - это болезнь избытка меди. Однако 95% меди в плазме переносится церулоплазмином, которого часто бывает мало при болезни Вильсона. Содержание меди в моче повышено при болезни Вильсона, и ее собирают в течение 24 часов в бутылке с вкладышем, не содержащим меди. Уровни выше 100 мкг / 24 ч (1,6 мкмоль / 24 ч) подтверждают болезнь Вильсона, а уровни выше 40 мкг / 24 ч (0,6 мкмоль / 24 ч) являются убедительным показателем.[5] Высокий уровень меди в моче не является уникальным признаком болезни Вильсона; они иногда наблюдаются в аутоиммунный гепатит И в холестаз (любое заболевание, препятствующее оттоку желчи от печени к тонкой кишке).[13]
У детей пеницилламин можно использовать тест. Вводят пероральную дозу пеницилламина 500 мг и собирают мочу в течение 24 часов. Если он содержит более 1600 мкг (25 мкмоль), это надежный индикатор болезни Вильсона.[требуется разъяснение ] Этот тест не прошел валидацию у взрослых.[13]
Биопсия печени
Как только другие исследования указали на болезнь Вильсона, идеальным тестом является удаление небольшого количества ткани печени через биопсия печени. Это оценивается микроскопически по степени стеатоз и цирроз, и гистохимия и количественное определение меди используются для измерения степени накопления меди. Уровень 250мкг меди на грамм высушенной ткани печени подтверждает болезнь Вильсона. Иногда обнаруживаются более низкие уровни меди; в этом случае комбинация результатов биопсии со всеми другими тестами все же может привести к формальному диагнозу Вильсона.[5]
На ранних стадиях заболевания биопсия обычно показывает: стеатоз (отложение жирового материала), повышенная гликоген в ядро, и области некроз (смерть клетки). При более запущенном заболевании наблюдаемые изменения очень похожи на изменения, наблюдаемые при аутоиммунном гепатите, например, инфильтрация воспалительный клетки, частичный некроз и фиброз (рубцовая ткань). Наконец, при запущенном заболевании главным признаком является цирроз. При острой печеночной недостаточности наблюдается дегенерация клеток печени и коллапс архитектуры ткани печени, как правило, на фоне цирротических изменений. Гистохимические методы обнаружения меди непоследовательны и ненадежны, и их использование по отдельности считается недостаточным для постановки диагноза.[13]
Генетическое тестирование
Мутационный анализ ATP7B ген, а также другие гены, связанные с накоплением меди в печени. После подтверждения мутации можно провести скрининг членов семьи на наличие болезни в рамках клиническая генетика семейное консультирование.[5] Важно следить за региональным распределением генов, связанных с болезнью Вильсона, поскольку это может помочь клиницистам разработать соответствующие стратегии скрининга. Поскольку мутации гена WD различаются в зависимости от популяции, исследования и генетическое тестирование, проводимые в таких странах, как США или Великобритания, могут создавать проблемы, поскольку они, как правило, имеют более смешанные популяции.[17]
Уход
Рацион питания
В целом рекомендуется диета с низким содержанием медьсодержащих продуктов, избегая грибы, орехи, шоколад сушеные фрукты, печень, семена кунжута и кунжутное масло, и моллюски.[5]
Медикамент
Для лечения болезни Вильсона доступны медицинские процедуры. Некоторые увеличивают выведение меди из организма, в то время как другие препятствуют усвоению меди из рациона.
В общем, пеницилламин это первое использованное лечение. Это связывает медь (хелатирование ) и приводит к выведению меди с мочой. Следовательно, можно контролировать количество меди в моче, чтобы гарантировать прием достаточно высокой дозы. Пеницилламин не обходится без проблем: около 20% испытывают побочный эффект или осложнение лечения пеницилламином, например, вызванное лекарством. волчанка (вызывая боли в суставах и кожную сыпь) или миастения (нервное состояние, приводящее к мышечной слабости). Почти половина из тех, у кого были неврологические симптомы, испытывают парадоксальное ухудшение симптомов. Хотя это явление наблюдается при других методах лечения Вильсона, его обычно принимают как показание для прекращения приема пеницилламина и начала лечения второй линии.[5][13] Лицам с непереносимостью пеницилламина можно вместо этого начать гидрохлорид триентина, который также обладает хелатирующими свойствами. Некоторые рекомендуют триентин в качестве лечения первой линии, но опыт применения пеницилламина более обширен.[13] Еще один агент, проходит клинические исследования Wilson Therapeutics, с известной активностью при болезни Вильсона тетратиомолибдат. Это считается экспериментальным,[13] хотя некоторые исследования показали положительный эффект.[5]
Как только все результаты вернутся к норме, цинк (обычно в виде ацетат цинка рецепт, называемый галзином) может использоваться вместо хелаторов для поддержания стабильного уровня меди в организме. Цинк стимулирует металлотионеин, белок в клетках кишечника, который связывает медь и предотвращает ее всасывание и транспортировку в печень. Терапию цинком продолжают, если симптомы не повторяются или если выведение меди с мочой не увеличивается.[13]
В редких случаях, когда никакие пероральные препараты не эффективны, особенно при тяжелых неврологических заболеваниях, димеркапрол (Британский анти-люизит) иногда бывает необходимо. Это лечение вводится внутримышечно (в мышцу) каждые несколько недель и имеет неприятные побочные эффекты, такие как боль.[18]
Люди, которые бессимптомный (например, те, кому поставлен диагноз в результате семейного скрининга или только в результате аномальных результатов анализов), как правило, лечат, поскольку накопление меди может вызвать долгосрочные повреждения в будущем. Неясно, лучше всего лечить этих людей пеницилламином или ацетатом цинка.[13]
Физическая и профессиональная терапия
Физиотерапия и трудотерапия полезны для пациентов с неврологической формой заболевания. Лечение хелатированием меди может занять до шести месяцев, и эти методы лечения могут помочь справиться с атаксия, дистония, и тремора, а также предотвращения развития контрактуры что может возникнуть в результате дистонии.[19]
Трансплантация
Трансплантация печени является эффективным лекарством от болезни Вильсона, но используется только в определенных сценариях из-за рисков и осложнений, связанных с процедурой. Он используется в основном у людей с молниеносный печеночная недостаточность, не поддающаяся лечению, или у пациентов с запущенным хроническим заболеванием печени. Трансплантации печени следует избегать при тяжелом нервно-психическом заболевании, при котором ее преимущества не были продемонстрированы.[5][13]
Прогноз
При отсутствии лечения болезнь Вильсона имеет тенденцию прогрессировать и в конечном итоге приводит к летальному исходу. При раннем выявлении и лечении большинство пострадавших могут жить относительно нормальной жизнью. Повреждения печени и неврологические нарушения, возникшие до лечения, могут улучшиться, но часто необратимы.[20]
История
Заболевание носит название Британский врач Сэмюэл Александр Киннер Уилсон (1878–1937), а невролог который описал состояние, в том числе патологические изменения в головном мозге и печени, в 1912 году.[21] Работе Уилсона предшествовали отчеты немецкого невролога. Карл Вестфаль (в 1883 г.), который назвал это «псевдосклерозом»; британский невролог Уильям Гауэрс (в 1888 г.);[22] финского невропатолога Эрнст Александр Хомен (в 1889-1892 гг.), отметившие наследственную природу болезни;[23] и по Адольф Струмпелл (в 1898 г.), отметившие цирроз печени.[22] Невропатолог Джон Натаниэль Камингс установил связь с накоплением меди как в печени, так и в мозге в 1948 году.[24] Возникновение гемолиза было отмечено в 1967 году.[25]
Камингс и одновременно новозеландский невролог Дерек Денни-Браун, работая в США, впервые сообщили об эффективном лечении хелатором металлов. Британский антилевизитский в 1951 г.[26][27] Это лечение нужно было вводить инъекциями, но это был один из первых методов лечения, доступных в области неврологии, области, которая классически могла наблюдать и диагностировать, но предлагала мало методов лечения.[22][28] Первый эффективный пероральный хелатирующий агент, пеницилламин, был открыт в 1956 году британским неврологом Джоном Уолшем.[29] В 1982 году Уолш также представил триентин,[30] и был первым, кто разработал тетратиомолибдат для клинического использования.[31] Терапия ацетатом цинка впервые появилась в Нидерландах, где врачи Скоувинк и Хугенрад использовали ее в 1961 и 1970-х годах, соответственно, но позже она получила дальнейшее развитие Брюэр и его коллеги. университет Мичигана.[18][32]
Генетическая основа болезни Вильсона и связь с ATP7B мутации были выявлены в 1980-х и 1990-х годах несколькими исследовательскими группами.[33][34]
Другие животные
Наследственное накопление меди описано в Бедлингтон-терьеры,[35] где это обычно влияет только на печень. Это связано с мутациями в COMMD1 (или же MURR1) ген.[36] Несмотря на эти выводы, COMMD1 мутации не могли быть обнаружены у людей с не вильсоновскими состояниями накопления меди (такими как Индийский цирроз в детстве ), чтобы объяснить их генетическое происхождение.[37]
Смотрите также
Рекомендации
- ^ а б c d е ж грамм час я «Болезнь Вильсона». NIDDK. Июль 2014 г. Архивировано с оригинал на 2016-10-04. Получено 2016-11-06.
- ^ Линн, Д. Джоанн; Ньютон, Герберт Б .; Рэй-Грант, Александр (2004). 5-минутная консультация невролога. Липпинкотт Уильямс и Уилкинс. п. 442. ISBN 9780683307238. В архиве из оригинала от 07.11.2016.
- ^ Sahani, Dushyant V .; Самир, Энтони Э. (2016). Абдоминальная визуализация: серия экспертных радиологов (2-е изд.). Elsevier Health Sciences. п. 400. ISBN 9780323431613. В архиве из оригинала от 07.11.2016.
- ^ «Whonamedit - словарь медицинских эпонимов». www.whonamedit.com. Архивировано из оригинал на 2016-11-07. Получено 2016-11-06.
- ^ а б c d е ж грамм час я j k л м п о п q р s т ты v Ала А., Уокер А.П., Ашкан К., Дули Дж.С., Шильский М.Л. (2007). «Болезнь Вильсона». Ланцет. 369 (9559): 397–408. Дои:10.1016 / S0140-6736 (07) 60196-2. PMID 17276780. S2CID 24663871.
- ^ а б c Мерл Ю., Шефер М., Ференци П., Стреммель В. (2007). «Клиническая картина, диагностика и отдаленные результаты болезни Вильсона: когортное исследование». Кишечник. 56 (1): 115–20. Дои:10.1136 / gut.2005.087262. ЧВК 1856673. PMID 16709660.
- ^ а б c Лоринц MT (2010). «Неврологическая болезнь Вильсона» (PDF). Летопись Нью-Йоркской академии наук. 1184 (1): 173–87. Bibcode:2010НЯСА1184..173Л. Дои:10.1111 / j.1749-6632.2009.05109.x. HDL:2027.42/78731. PMID 20146697. S2CID 2989668.
- ^ Пагонабаррага, Дж; Гетц, С. (2012). Биллер, Дж (ред.). Практическая неврология (4-е изд.). Филадельфия: Уолтерс Клувер / Липпинкотт Уильямс и Уилкинс Хит. п. 282. ISBN 978-1451142631.
- ^ Янофф, Мирон; Джей С. Дукер (2008). Офтальмология (3-е изд.). Эдинбург: Мосби. п. 411. ISBN 978-0323057516.
- ^ а б c d е ж грамм де Би П., Мюллер П., Вейменга С., Кломп Л. В. (ноябрь 2007 г.). «Молекулярный патогенез болезни Вильсона и Менкеса: корреляция мутаций с молекулярными дефектами и фенотипами болезни». J. Med. Genet. 44 (11): 673–88. Дои:10.1136 / jmg.2007.052746. ЧВК 2752173. PMID 17717039.
- ^ Кумар, Мукеш; Гахарвар, Уткарш; Пол, Сангита; Пуджари, Мукта; Пандхаре, Кавита; Скария, Винод; Бк, Бинукумар (2020-06-03). "WilsonGen - исчерпывающий ресурс с клиническими аннотациями геномных вариантов болезни Вильсона". Научные отчеты. 10 (1): 9037. Bibcode:2020НатСР..10.9037K. Дои:10.1038 / s41598-020-66099-2. ISSN 2045-2322. ЧВК 7270127. PMID 32493955.
- ^ Grubenbecher S, Stüve O, Hefter H, Korth C (2006). «Кодон 129 гена прионного белка модулирует клиническое течение неврологической болезни Вильсона». NeuroReport. 17 (5): 549–52. Дои:10.1097 / 01.wnr.0000209006.48105.90. PMID 16543824. S2CID 37186426.
- ^ а б c d е ж грамм час я j k л м Робертс, Ева А .; Шильский, Майкл Л. (2003). «Практическое руководство по болезни Вильсона» (PDF). Гепатология. 37 (6): 1475–92. Дои:10.1053 / jhep.2003.50252. PMID 12774027. S2CID 263620.[мертвая ссылка ]
- ^ Ли Г.Р. (1999). «Глава 48: приобретенные гемолитические анемии, возникшие в результате прямого воздействия инфекционных, химических или физических агентов». В Lee GR, Foerster J, Lukens J и др. (ред.). Клиническая гематология Винтроба. том 1 (10-е изд.). Уильямс и Уилкинс. стр.1298. ISBN 978-0-683-18242-2.
- ^ Шейвер WA, Бхатт H, Combes B (1986). «Низкая активность щелочной фосфатазы в сыворотке при болезни Вильсона». Гепатология. 6 (5): 859–63. Дои:10.1002 / hep.1840060509. PMID 3758940. S2CID 24055787.
- ^ Das SK, Ray K (сентябрь 2006 г.). «Болезнь Вильсона: обновление». Нат Клин Прак Нейрол. 2 (9): 482–93. Дои:10.1038 / ncpneuro0291. PMID 16932613. S2CID 205340375.
- ^ Ференчи, Питер (22.06.2006). «Региональное распределение мутаций гена ATP7B у пациентов с болезнью Вильсона: влияние на генетическое тестирование». Генетика человека. 120 (2): 151–159. Дои:10.1007 / s00439-006-0202-5. ISSN 0340-6717. PMID 16791614. S2CID 10124665.
- ^ а б Уолше Дж. М. (июль 1996 г.). «Лечение болезни Вильсона: историческая справка». QJM. 89 (7): 553–5. Дои:10.1093 / qjmed / 89.7.553. PMID 8759497.
- ^ Брюэр Г.Дж., Аскари Ф.К. (2005). «Болезнь Вильсона: клиническое ведение и терапия». Журнал гепатологии. 42 (Дополнение 1): 13–21. Дои:10.1016 / j.jhep.2004.11.013. PMID 15777568.
- ^ «Определение и факты | NIDDK». Национальный институт диабета, болезней органов пищеварения и почек. Получено 2019-02-01.
- ^ Киннер Вильсон С.А. (1912). «Прогрессирующая линзовидная дегенерация: семейное нервное заболевание, связанное с циррозом печени». Мозг. 34 (1): 295–507. Дои:10.1093 / мозг / 34.4.295. Архивировано из оригинал (PDF) на 2009-09-03. Получено 2008-04-09.
- ^ а б c Робертсон WM (февраль 2000 г.). «Болезнь Вильсона». Arch. Neurol. 57 (2): 276–7. Дои:10.1001 / archneur.57.2.276. PMID 10681092.
- ^ Homén EA (1892). "Eine eigenthümliche bei drei Geschwistern auftretende typische Krankheit unter der Form einer прогрессивная деменция в Verbindung mit ausgedehnten Gefässveränderungen (wohl Lues hereditaria tarda)". Archiv für Psychiatrie und Nervenkrankheiten. 24: 1–38.
- ^ Камингс Дж. Н. (1948). «Содержание меди и железа в головном мозге и печени при нормальной и гепато-линзовидной дегенерации». Мозг. 71 (Декабрь): 410–5. Дои:10.1093 / мозг / 71.4.410. PMID 18124738. Архивировано из оригинал (PDF) на 2009-09-03. Получено 2008-04-09.
- ^ Макинтайр Н., Клинк Х.М., Леви А.Дж., Камингс Дж. Н., Шерлок С. (февраль 1967 г.). «Гемолитическая анемия при болезни Вильсона». N. Engl. J. Med. 276 (8): 439–44. Дои:10.1056 / NEJM196702232760804. PMID 6018274.
- ^ Камингс Дж. Н. (март 1951 г.). «Эффекты Б.А.Л. при гепатолентикулярной дегенерации». Мозг. 74 (1): 10–22. Дои:10.1093 / мозг / 74.1.10. PMID 14830662.
- ^ Денни-Браун Д., Портер Х (декабрь 1951 г.). «Влияние БАЛ (2,3-димеркаптопропанол) на гепатолентикулярную дегенерацию (болезнь Вильсона)». N. Engl. J. Med. 245 (24): 917–25. Дои:10.1056 / NEJM195112132452401. PMID 14882450.
- ^ Виленский JA, Робертсон WM, Гилман S (сентябрь 2002 г.). «Денни-Браун, болезнь Вильсона и БАЛ (британский антилевизит [2,3-димеркаптопропанол])». Неврология. 59 (6): 914–6. Дои:10.1212 / wnl.59.6.914. PMID 12297577.
- ^ Уолше Дж. М. (январь 1956 г.). «Болезнь Вильсона; новая пероральная терапия». Ланцет. 270 (6906): 25–6. Дои:10.1016 / S0140-6736 (56) 91859-1. PMID 13279157.
- ^ Уолше Дж. М. (март 1982 г.). "Лечение болезни Вильсона дигидрохлоридом триентина (триэтилентетрамина)". Ланцет. 1 (8273): 643–7. Дои:10.1016 / S0140-6736 (82) 92201-2. PMID 6121964. S2CID 205999334.
- ^ Харпер П.Л., Уолше Дж. М. (декабрь 1986 г.). «Обратимая панцитопения, вызванная лечением тетратиомолибдатом». Br. J. Haematol. 64 (4): 851–3. Дои:10.1111 / j.1365-2141.1986.tb02250.x. PMID 3801328. S2CID 11546705.
- ^ Брюэр Дж. Дж. (Январь 2000 г.). «Распознавание, диагностика и лечение болезни Вильсона». Proc. Soc. Exp. Биол. Med. 223 (1): 39–46. Дои:10.1046 / j.1525-1373.2000.22305.x. PMID 10632959. Архивировано из оригинал на 2008-04-09. Получено 2008-05-20.
- ^ Bull PC, Томас Г.Р., Ромменс Дж. М., Форбс Дж. Р., Кокс Д. В. (1993). «Ген болезни Вильсона - это предполагаемая АТФаза Р-типа, транспортирующая медь, похожая на ген Менкеса». Nat. Genet. 5 (4): 327–37. Дои:10.1038 / ng1293-327. PMID 8298639. S2CID 1236890.
- ^ Танзи Р.Е., Петрухин К., Чернов И. и др. (1993). «Ген болезни Вильсона представляет собой АТФазу, транспортирующую медь, гомологичную гену болезни Менкеса». Nat. Genet. 5 (4): 344–50. Дои:10.1038 / ng1293-344. PMID 8298641. S2CID 610188.
- ^ Sternlieb I, Twedt DC, Johnson GF и др. (1977). «Унаследованная медная токсичность печени у бедлингтонских терьеров». Proc. R. Soc. Med. 70 Дополнение 3 (Дополнение 3): 8–9. ЧВК 1543595. PMID 122681.
- ^ ван Де Слуис Б., Ротуйзен Дж., Пирсон П.Л., ван Ост Б.А., Вейменга С. (2002). «Идентификация нового гена метаболизма меди путем позиционного клонирования в популяции чистокровных собак». Гм. Мол. Genet. 11 (2): 165–73. Дои:10.1093 / hmg / 11.2.165. PMID 11809725. Архивировано из оригинал на 2008-08-30. Получено 2008-04-11.
- ^ Мюллер Т., ван де Слуис Б., Жернакова А. и др. (2003). «Ген токсикоза меди у собак MURR1 не вызывает невильсоновского токсикоза меди в печени». J. Hepatol. 38 (2): 164–8. Дои:10.1016 / S0168-8278 (02) 00356-2. PMID 12547404.
внешняя ссылка
- Болезнь Вильсона в Керли
- Болезнь Вильсона в NLM Домашний справочник по генетике
| Классификация | |
|---|---|
| Внешние ресурсы |