Общий синтез витамина B12 - Vitamin B12 total synthesis
Эта статья может содержать чрезмерное количество сложных деталей, которые могут заинтересовать только определенную аудиторию. (Июнь 2020 г.) (Узнайте, как и когда удалить этот шаблон сообщения) |
В полный синтез сложной биомолекулы витамин B12 было выполнено двумя разными подходами совместными исследовательскими группами Роберт Бернс Вудворд в Гарвард[1][2][3][4][5] и Альберт Эшенмозер в ETH[6][7][8][9][10][11][12] в 1972 году. Это достижение потребовало усилий не менее 91 человека. постдокторанты (Гарвард: 77, ETH: 14)[13]:9-10[14], и 12 докторов наук. студенты (в ETH[12]:1420) из 19 разных стран в течение почти 12 лет.[5](1:14:00-1:14:32,1:15:50-1:19:35)[14]:17-18 Проект синтеза[15] вызвало и повлекло за собой серьезные изменения парадигма[16][17]:37[18]:1488 в области натуральный продукт синтез.[19][20][21]
Молекула

Витамин B12, С63ЧАС88Против14О14P, является наиболее сложным из всех известных витамины. Его химическая структура была определена рентгеновский анализ кристаллической структуры в 1956 г. исследовательской группой Дороти Ходжкин (Оксфордский университет ) в сотрудничестве с Кеннет Н. Трублад в UCLA и Джон Г. Уайт в Университет Принстона.[22][23]Ядро молекулы - это Коррин структура, азотистая тетрадентатный лиганд система.[примечание 1] Это биогенетически относится к порфирины и хлорофиллы, но отличается от них в важных отношениях: в углеродном скелете отсутствует один из четырех мезоуглеродов между пятичленными кольцами, причем два кольца (A и D, рис. 1) напрямую соединены одинарная связь углерод-углерод. Коррин хромофор Таким образом, система является нециклической и расширяется только на три мезопозиции, включая три винилогичный амидин единицы. Выстроились на периферии макроцикл кольцо восемь метил группы и четыре пропионовый и три уксусная кислота боковые цепи. Девять атомов углерода на периферии коррина хирогенные центры. Тетрадентатная, одноосновный лиганд коррина экваториально согласованный с талантом кобальт ион, который несет два дополнительных осевой лиганды.[заметка 2]

Несколько натуральных вариантов B12 Существуют структуры, которые различаются этими аксиальными лигандами. В самом витамине кобальт несет циано группа на верхней стороне коррина плоскости (цианокобаламин ), а нуклеотид петля с другой. Эта петля соединена на другом конце с периферической пропионовой амидной группой в кольце D и состоит из структурных элементов, полученных из аминопропанол, фосфат, рибоза, и 5,6-диметилбензимидазол. Один из атомов азота имидазол кольцо аксиально координировано с кобальтом, таким образом, нуклеотидная петля образует девятнадцатичленное кольцо. Все карбоксильные группы боковых цепей являются амидами.
Кобириновая кислота, одно из природных производных витамина B12,[24] отсутствует нуклеотидная петля; в зависимости от природы двух аксиальных лигандов, вместо этого он проявляет свою функцию пропионовой кислоты в кольце D в виде карбоксилата (как показано на фиг.1) или карбоновой кислоты (с двумя цианидными лигандами у кобальта).
Два синтеза
Структура витамин B12 был первым низкомолекулярным натуральный продукт определяется рентгеновским анализом, а не химическим разложением. Таким образом, пока структура этого нового типа комплекса биомолекула был установлен, его химический состав остался по существу неизвестным; исследование этой химии стало одной из задач витаминного химический синтез.[12]:1411[18]:1488-1489[25]:275 В 1960-х годах синтез такой исключительно сложной и уникальной структуры представлял собой серьезную проблему на переднем крае исследований в области синтеза органических природных продуктов.[17]:27-28[1]:519-521


Уже в 1960 году исследовательская группа биохимика Конрад Бернхауэр в Штутгарт восстановил витамин B12 из одного из его производных в природе, кобировой кислоты,[24] поэтапным построением витаминной нуклеотидной петли.[примечание 4] Эта работа составила частичный синтез витамина B12 из натурального продукта, содержащего все структурные элементы витамина B12 кроме нуклеотид петля. Поэтому кобириновая кислота была выбрана в качестве целевой молекулы для полного синтеза витамина B.12.[6]:183-184[1]:521[8]:367-368
Совместная работа[3]:1456[17][28]:302-313 исследовательских групп на Гарвард и в ETH привели к двум синтезу кобировой кислоты, оба одновременно завершены в 1972 году,[29][30] один в Гарварде[3], а другой - в ETH.[10][11][12] «Конкурентное сотрудничество»[17]:30[31]:626 такого размера, включая 103 аспиранта и докторанта, в общей сложности почти 177 человеко-лет,[13]:9-10 пока что уникален в истории органический синтез.[4](0:36:25-0:37:37) Эти два синтеза сложно химически переплетены,[18]:1571 но они различаются в основном способом макроцикл Построена коррин-лигандная система. Обе стратегии построены по образцу двух модельных синтезов коррин, разработанных в ETH.[8][18]:1496,1499[32]:71-72 Первый, опубликованный в 1964 г.,[26] достигли конструкции хромофора коррина путем объединения A-D-компонента с B-C-компонентом через иминоэстер /енамин -C, C-конденсаты, при этом окончательное замыкание корринговых колец достигается между кольцами A и B.[33] Второй синтез модели, опубликованный в 1969 г.,[34] исследовал роман фотохимический процесс циклоизомеризации для создания прямого соединения A / D-кольца в качестве окончательного замыкания коррин-кольца между кольцами A и D.[35]
Подход A / B к синтезу кобириновой кислоты был совместно разработан и реализован в 1972 году в Гарварде. Он сочетал в себе бициклический Гарвардский компонент A-D с ETH B-C-компонент, и замкнул макроциклическое корриновое кольцо между кольцами A и B.[3]:145,176[4](0:36:25-0:37:37) Подход A / D к синтезу, реализованный в ETH и завершенный одновременно с подходом A / B также в 1972 году, последовательно добавляет: кольца D и A к B-C-компоненте подхода A / B и выходит на корринговое кольцо замыкание между кольцами A и D.[10][11][12] Пути этих двух синтезов встретились в общем корриноидном промежуточном продукте.[11]:519[36]:172 В последние шаги превращение этого промежуточного продукта в кобировую кислоту были проведены в двух лабораториях снова совместно, каждая группа работала с материалом, приготовленным с помощью своего собственного подхода, соответственно.[17]:33[18]:1567
Краткий обзор сотрудничества Гарварда и ETH
Начала
Woodward и Eschenmoser приступил к проекту химического синтеза витамина B12 независимо друг от друга. Группа ETH начала с модельного исследования того, как синтезировать систему лигандов коррина в декабре 1959 года.[18]:1501 В августе 1961 г.[17]:29[13]:7 группа Гарварда начала атаковать наращивание B12 структуру напрямую, нацеливаясь на наиболее сложную часть B12 молекула, "западная половина"[1]:539 который содержит прямое соединение между кольцами A и D (A-D-компонент). Уже в октябре 1960 г.[17]:29[13]:7[37]:67 группа ETH начала синтез предшественника витамина B с кольцом B12.
В начале,[38] Прогресс в Гарварде был быстрым, пока неожиданный стереохимический ход стадии формирования центрального кольца не прервал проект.[39][17]:29 Признание Вудворда стереохимической загадки, обнаруженной раздражающим поведением одного из его тщательно спланированных синтетических шагов, стало, согласно его собственным сочинениям,[39] часть событий, которые привели к правила орбитальной симметрии.
После 1965 года группа из Гарварда продолжила работу в направлении A-D-компонент по измененному плану, используя (-) - камфора[40] как источник кольца D.[17]:29[18]:1556
Объединение усилий: подход A / B к синтезу кобириновой кислоты
К 1964 году группа ETH выполнила первый Коррин синтез модели,[26][25]:275 а также получение предшественника кольца B как часть конструкции B12 сама молекула.[37][41] Поскольку независимый прогресс двух групп в достижении их долгосрочной цели явно дополнял друг друга, Вудворд и Эшенмозер решили в 1965 г.[18]:1497[17]:30 объединить усилия и с тех пор продолжать проект B12 синтез совместно, планируя использовать стратегию конструирования лиганда (кольцевого соединения компонентов) модельной системы ETH.[2]:283[18]:1555-1574
К 1966 году группе ETH удалось синтезировать B-C-компонент («восточная половина»[1]:539) путем связывания их предшественника кольца-B с предшественником кольца-C.[18]:1557 Последний также был приготовлен в Гарварде из (-) - камфоры с помощью стратегии, разработанной и использованной ранее А. Пелтером и Дж. У. Корнфорт в 1961 г.[примечание 6] В ETH синтез B-C-компонента включал реализацию реакции C, C-конденсации через сульфидное сжатие. Оказалось, что этот недавно разработанный метод обеспечивает общее решение проблемы построения характерных структурных элементов хромофора коррина - винилогичных амидиновых систем, соединяющих четыре периферических кольца.[18]:1499
В начале 1967 года группа Гарварда завершила синтез модельного A-D-компонента,[примечание 7] с недифференцированной f-боковой цепью, несущей функцию сложного метилового эфира, как и все другие боковые цепи.[18]:1557 С этого момента две группы систематически обменивались образцами соответствующих половин целевой структуры корриноида.[17]:30-31[18]:1561[30]:17 К 1970 году они совместно соединили недифференцированный A-D-компонент Гарварда с B-C-компонентом ETH, получив дицианокобальт (III) -5,15-биснор-гептаметил-кобиринат. 1 (рис.4).[заметка 2] Группа ETH определила этот полностью синтетический корриноидный промежуточный продукт путем прямого сравнения с образцом, полученным из натурального витамина B.12.[2]:301-303[18]:1563
В этом усовершенствованном модельном исследовании условия реакции для требовательных процессов C / D-соединение и A / B-циклизация методом сульфидного сжатия. Условия C / D-соединения были успешно исследованы в обеих лабораториях, лучшие условия были найдены в Гарварде,[2]:290-292[18]:1562 в то время как метод замыкания A / B-кольца через внутримолекулярный версия сульфидное сжатие[44][34][45] был разработан в ETH.[2]:297-299[46][18]:1562-1564 Позже в Гарварде было показано, что замыкание A / B-кольца также может быть достигнуто с помощью тио-иминоэфирная / енаминная конденсация.[2]:299-300[18]:1564
К началу 1971 года группа Гарварда завершила синтез последнего A-D-компонента,[примечание 8] содержащие карбоксильную функцию f-боковой цепи в кольце D, отличную от всех карбоксильных функций как нитрильную группу (как показано на 2 в инжир. 4; см. также рис. 3 ).[3]:153-157 A / D-часть B12 структура включает в себя наиболее сложную по конституции и конфигурации часть молекулы витамина; его синтез рассматривается как апофеоз искусства Вудвардиана в полном синтезе натуральных продуктов.[11]:519[12]:1413[18]:1564[31]:626
Альтернативный подход к синтезу кобирной кислоты
Еще в 1966 году[35]:1946 группа ETH начала исследовать, снова в модельной системе, альтернативную стратегию синтеза коррина, в которой кольцо коррина будет замкнуто между кольцами A и D. Проект был вдохновлен возможным существованием пока неизвестной реоганизации связей. процесс.[35]:1943-1946 Это, если таковое существует, сделало бы возможным создание кобировой кислоты из одного исходного материала.[6]:185[8]:392,394-395[31] Важно отметить, что гипотетический процесс, интерпретируемый как подразумевающий две последовательные перегруппировки, был признан формально охватываемым новыми классификациями реактивности сигматропных перегруппировок и электроциклизаций, предложенными Woodward и Hoffmann в контексте их правила орбитальной симметрии![8]:395-397,399[11]:521[47][18]:1571-1572
К маю 1968 г.[18]:1555 Группа ETH продемонстрировала в модельном исследовании, что предусмотренный процесс, фотохимическая циклоизомеризация A / D-секокорринат → корринат, действительно существует. Впервые было обнаружено, что этот процесс протекает с комплексом Pd, но не с соответствующими комплексами Ni (II) - или кобальта (III) -A / D-секокорринат.[34][48]:21-22 Это также плавно происходило в комплексах ионов металлов, таких как цинк, и других фотохимически инертных и слабосвязанных ионов металлов.[8]:400-404[12]:1414 После закрытия кольца их можно было легко заменить кобальтом.[8]:404 Эти открытия открыли дверь к тому, что в конечном итоге стало фотохимический A / D подход синтеза кобирной кислоты.[7]:31[9]:72-74[35]:1948-1959

С осени 1969 г.[49]:23 с B-C-компонент подхода A / B и предшественника кольца D, приготовленного из энантиомер Из исходного материала, ведущего к предшественнику кольца B, потребовался аспирант Вальтер Фюрер[49] менее полутора лет[17]:32 трансформировать фотохимическую модель синтеза коррина в синтез дицианокобальта (III) -5,15-биснор-a, b, d, e, g-пентаметил-кобиринат-c-N, N-диметиламид-f-нитрил 2 (инжир. 4 ), обычный промежуточный корриноид на пути к кобировой кислоте. В Гарварде тот же средний 2 был получен примерно в то же время путем соединения дифференцированного кольцевого D-дифференцированного Гарвардского A-D-компонента (доступен весной 1971 г.[18]:Сноска 54a 1564[3]:153-157) с B-C-компонентом ETH, применяя методы конденсации, разработанные ранее с использованием недифференцированной A-D-компоненты.[1]:544-547[2]:285-300
Таким образом, весной 1971 г.[31]:634 два разных пути к обычному промежуточному корриноиду 2 (инжир. 4 ) на пути к кобириновой кислоте, для чего потребовалось 62 химических этапа (Гарвард / ETH A / B подход ), остальные 42 (ETH A / D подход ). В обоих подходах четыре периферийных кольца, полученные из энантиочистка предшественники, обладающие правильным чувством хиральный, тем самым обходя основные стереохимические проблемы при накоплении лигандной системы.[1]:520-521[7]:12-13[11]:521-522 При построении A / D-перехода A / D-секокоррином →Коррин циклоизомеризация, образование двух A / D-диастереомеры следовало ожидать. Использование кадмия (II) в качестве координирующего иона металла привело к очень высокой диастереоселективности.[49]:44-46 в пользу натурального A / D-транс-изомер.[12]:1414-1415
Как только структура коррина была сформирована любым подходом, три C-H-хирогенные центры на периферии, прилегающей к хромофор система оказалась склонной к эпимеризация с исключительной легкостью.[2]:286[9]:88[3]:158[4](1:53:33-1:54:08)[18]:1567 Это потребовало разделения диастереомеров после большинства химических стадий на этой продвинутой стадии синтезов. Действительно, к счастью, примерно в то время техника жидкостная хроматография высокого давления (ВЭЖХ) были разработаны в аналитической химии.[50] ВЭЖХ стала незаменимым инструментом в обеих лабораториях;[30]:25[9]:88-89[3]:165[4](0:01:52-0:02:00,2:09:04-2:09:32) его использование в B12 проект, инициированный Якобом Шрайбером из ETH,[51] был первым применением этой техники в синтезе натуральных продуктов.[18]:1566-1567[36]:190[52]
Совместные заключительные шаги
В окончательное преобразование обыкновенного промежуточного корриноида 2 (рис. 6) из двух подходов в целевую кобировую кислоту потребовалось введение двух недостающих метильные группы в мезоположениях хромофора коррина между кольцами A / B и C / D, а также преобразование всех периферических карбоксильных функций в их амидную форму, за исключением критического карбоксила в кольцевой f-боковой цепи D (см. рис. 6). Эти этапы были совместно исследованы строго параллельно в обеих лабораториях: группа Гарварда с использованием материала, полученного с помощью подхода A / B, группа ETH, подготовленная с помощью фотохимического подхода A / D.[17]:33[18]:1567

Первое решительное отождествление полностью синтетический средний на пути к кобирной кислоте был проведен в феврале 1972 г. с кристаллическим образцом полностью синтетического дицианокобальта (III) -гексаметил-кобиринат-f-амида. 3 (рис.6[заметка 2]), который во всех данных оказался идентичным кристаллическому релейному образцу, сделанному из витамина B.12 метанолизом до кобэстера 4,[примечание 9] с последующим частичным аммонолизом и разделением полученной смеси.[53]:44-45,126-143[3]:170[55]:46-47 В то время, когда Вудворд объявил «Полный синтез витамина B»12"на конференции ИЮПАК в Нью-Дели в феврале 1972 г.,[3]:177 полностью синтетический образец был приготовлен в ETH с помощью фотохимического A / D подхода,[17]:35[56]:148[18]:1569-1570 в то время как первый образец синтетической кобировой кислоты, идентифицированной с природной кобировой кислотой, был изготовлен в Гарварде, начиная с Б.12-производный ф-амидный материал реле.[55]:46-47[3]:171-176 Таким образом, достижением Вудворда / Эшенмозера в то время было, строго говоря, два формальных полного синтеза кобириновой кислоты, а также формальный тотальный синтез витамина.[55]:46-47[18]:1569-1570
Позднее, в 1972 г., два кристаллических эпимеры полностью синтетического дициано-кобальта (III) -гексаметил-кобирината-f-амид 3, а также два кристаллических эпимера полностью синтетического f-нитрила, полученные обоими способами синтеза, были строго идентифицированный хроматографически и спектроскопически с соответствующим B12-производные вещества.[18]:1570-1571[53]:181-197,206-221[5](0:21:13-0:46:32,0:51:45-0:52:49)[57] Затем в Гарварде кобировую кислоту производили также из полностью синтетического ф-амида. 3 подготовлены с использованием подхода A / B.[55]:48-49 Наконец, в 1976 году в Гарварде[55] полностью синтетическая кобирная кислота была преобразована в витамин B12 по пути, открытому Конрад Бернхауэр.[примечание 4]
Запись о публикации
В течение почти 12 лет, которые потребовались обеим группам для достижения своей цели, и Вудворд, и Эшенмозер периодически докладывали о стадии совместного проекта в виде лекций, некоторые из которых появлялись в печати. Woodward обсудили подход A / B в лекциях, опубликованных в 1968 г.,[1] и 1971 г.,[2] кульминацией стало объявление «Всего синтеза витамина B»12"в Нью-Дели в феврале 1972 г.[3]:177 опубликовано в 1973 г.[3] Эта публикация и лекции под тем же названием Вудворд прочитали в конце 1972 года.[4][5] ограничиваются подходом A / B синтеза и не обсуждают подход ETH A / D.
Eschenmoser обсудили вклад ETH в подход A / B в 1968 году на 22-м Фонд Роберта А. Велча конференция в Хьюстоне,[7] а также в его 1969 г. Лекция к столетию РНЦ "Дороги к Корринсу", опубликовано в 1970 году.[8] Он представил фотохимический A / D подход ETH к B12 синтез на 23-м ИЮПАК Конгресс в Бостоне в 1971 году.[9] Цюрихская группа объявила о завершении синтеза кобириновой кислоты фотохимическим A / D-подходом в двух лекциях, прочитанных аспирантами Маагом и Фюрером на заседании Швейцарского химического общества в апреле 1972 года.[10] Эшенмозер представил лекцию «Полный синтез витамина B».12: фотохимический путь »впервые в качестве лекции Уилсона Бейкера в Бристольском университете, Бристоль / Великобритания, 8 мая 1972 года.[примечание 10]


В качестве совместной полной публикации синтезов групп Гарварда и ETH (объявлено в[10] и ожидается в[11]) не появилось к 1977 г.,[примечание 12] статья, описывающая окончательную версию фотохимического A / D подхода, уже реализованную в 1972 г.[10][49][53][61] был опубликован в 1977 году в журнале Science.[12][56]:148 Эта статья представляет собой расширенный перевод на английский язык статьи, которая уже была опубликована в 1974 г. в Naturwissenschaften,[11] на основе лекции, прочитанной Эшенмозером 21 января 1974 г. на собрании Zürcher Naturforschende Gesellschaft. Четыре десятилетия спустя, в 2015 году, тот же автор наконец опубликовал серию из шести полных статей, описывающих работу группы ETH по Коррин синтез.[62][18][63][64][33][35] Часть I серии содержит главу, озаглавленную «Заключительный этап сотрудничества Гарварда и ETH по синтезу витамина B».12",[18]:1555-1574 в котором вклад группы ETH в совместную работу по синтезу витамина B12 между 1965 и 1972 годами.
Целиком ETH Работа подробно описана экспериментально в общедоступном документе Ph.D. диссертации,[37][41][58][44][59][54][60][42][46][49][53][61] почти 1'900 страниц, все на немецком языке.[65] Вклады 14 постдокторских исследователей ETH, участвовавших в синтезе кобириновой кислоты, в основном объединены в эти тезисы.[12]:1420[62]:1480[13]:12,38 Подробная экспериментальная работа на Гарвард был задокументирован в отчетах 77 исследователей, получивших докторскую степень, общим объемом более 3000 страниц.[13]:9,38[примечание 11]
Репрезентативные обзоры двух подходов к химическому синтезу витамина B12 были подробно опубликованы А. Х. Джексоном и К. М. Смитом,[43] Т. Гото,[66] Р. В. Стивенс,[36] К. К. Николау И Э. Г. Соренсен,[15][19] резюмировано Дж. Мулзер И Д. Ритер,[67] и Г. В. Крейг,[14][31] помимо многих других публикаций, в которых обсуждаются эти эпохальные синтезы.[примечание 13]
Подход Гарварда / ETH к синтезу кобировой кислоты: путь к обычному промежуточному корриноиду через замыкание A / B-корринового кольца
В подходе A / B к кобириновой кислоте гарвардский A-D-компонент был связан с ETH. B-C-компонент между кольцами D и C, а затем замкнулся в коридор между кольцами A и B. Оба этих критических шага были выполнены с помощью C, C-соединение посредством сульфидного сжатия, новый тип реакции, разработанный в синтезе B-C-компонента в ETH. Компонент A-D был синтезирован в Гарварде из предшественника кольца A (полученного из ахиральный исходные материалы), а также предшественник кольца D, полученный из (-) - камфора. Модель A-D-компонента использовалась для исследования условий сцепления; этот компонент отличается от компонента A-D, используемого в конечном синтезе, тем, что в качестве функциональной группы в боковой цепи f кольца D имеется метиловый эфир группу (как и все другие боковые цепи) вместо нитрил группа.
| Гарвардский синтез A-D-компонентов для A / B-подхода |
|---|
Синтез предшественника кольца-А Рисунок 8: Гарвардский синтез A-D-компонентов: кольцо A Отправной точкой для синтеза предшественника кольца A был метоксидиметилиндол. H-1 синтезировано конденсация из База Шиффа из м-анизидин и ацетоин. Реакция с Реактив Гриньяра из пропаргил йодид дал рацемический пропаргил индоленин гонка-H-2; закрытие кольца на аминокетон гонка-H-3 был вызван BF3 и HgO в MeOH через промежуточный гонка-H-2a (электрофильный добавление) с двумя метильными группами, вынужденными СНГ-зависимость как по кинетическим, так и по термодинамическим причинам.[1]:521-522  Рисунок 9: Гарвардский синтез A-D-компонентов: разрешение кольца A Разрешение рацемического аминокетона на два энантиомеры. Реакция гонка-H-3 с (-) - этилом изоцианат разрешенная изоляция кристаллизация одного из двух диастереомерный образуются производные мочевины (другая не кристаллизуется). Обработка рацемического кетона гонка-H-3 (или из маточные жидкости от предыдущей кристаллизации) с (+) - этилизоцианатом дал энантиомер первого мочевина производная. Пиролитический разложение каждого из этих производных мочевины привело к энантиочистка аминокетоны, желаемые (+) - Н-3, и (-) - Н-3.[1]:524-525 «Неестественный» (-) - энантиомер (-) - Н-3 использовался для определения абсолютного конфигурация; на различных последующих этапах, (-) - Н-3 и производные от него энантио-промежуточные соединения были использованы в качестве модельных соединений в поисковых экспериментах.[36]:173 Woodward написал о неестественном энантиомере, «наш опыт таков, что это почти единственный вид модельного исследования, который мы считаем полностью надежным».[1]:529  Рисунок 10: Гарвардский синтез A-D-компонентов: определение конфигурации кольца A Определение абсолютная конфигурация предшественника кольца A (+) - H-3. Для этого определения левовращающий («неестественный») энантиомер аминокетона (-) - Н-3 был использован для сохранения драгоценного материала: ацилирование аминогруппы (-) - Н-3 с хлорацетилхлорид с последующей обработкой продукта H-3a с калий т-бутоксид в т-бутанол, дает тетрациклический кето-лактам H-3b. Его кетокарбонил был преобразован в метиленовую группу посредством обессеривание из дитиокетальный из H-3b с Никель Ренея давать лактам H-3c. Разрушение ароматического кольца озонолиз, включая потерю карбоксильной функции спонтанной декарбоксилирование, привело к бициклической лактамкарбоновой кислоте H-3d. Этот материал был идентифицирован как продукт H-3h происходит от (+) - камфора, имеющий такое же строение и абсолютную конфигурацию, как показано в формуле H-3d.[1]:525-526 Материал для идентификации H-3d был синтезирован из (+) - камфоры следующим образом: СНГ-изокетопиновая кислота H-3e, полученный из (+) - камфары установленным путем, описанным в литературе,[68] был преобразован через соответствующий хлористый, азид, и изоцианат к метил-уретан H-3f. При лечении калием т-бутоксид в т-бутанол, а затем КОН, H-3f был преобразован в H-3h, явно через промежуточный H-3g. Идентичность двух образцов H-3d и H-3h полученный двумя описанными способами, установил абсолютную конфигурацию (+) - Н-3, энантиомер предшественника кольца-A.[1]:525-526 Синтез предшественника кольца D из (-) - камфоры  Рисунок 11: Гарвардский синтез A-D-компонентов: кольцо D из (-) - камфоры (-) - Камфора была нитрозированный в α-положении карбонильной группы, чтобы дать оксим H-4, Декольте Бекмана образуется через соответствующий нитрил амид H-5. Деградация Гофмана через промежуточный амин и его замыкание в кольцо привело к лактаму H-6. Преобразование своего N-нитрозопроизводная H-7 дал диазо сложный H-8. Термическое разложение H-8 индуцированный метил миграция дать циклопентен H-9. Приведение к H-10 (LiAlH4 ), окисление (хромовая кислота ) в альдегид H-11, Реакция Виттига (карбометоксиметилентрифенилфосфоран ) к H-12 и гидролиз сложноэфирной группы окончательно дал транс-карбоновая кислота H-13.[1]:527-528[примечание 14] Сочетание предшественников кольца A и кольца D с «пентацикленоном»  Рисунок 12: Гарвардский синтез A-D-компонентов: соединение колец A и D с «пентациленоном» N-ацилирование трициклического аминокетона (+) - Н-3 с хлоридом H-14 карбоновой кислоты H-13 дал амид H-15, который при лечении калием т-бутоксид в т-бутанол стереоселективно произведенный пентациклический кето-лактам H-16 через внутримолекулярный Реакция Майкла который направляет указанные атомы водорода в транс-отношении друг к другу. В ожидании Сокращение березы из ароматный кольцо, защитные группы для двоих карбонильные функции из H-16 потребовались, один для карбонильной группы кетона как кеталь H-17, а другой для лактам карбонил как высокочувствительный енольный эфир H-20. Последний защита был достигнут лечением H-17 с Соль Меервейна (тетрафторборат триэтилоксония) давать иминиевая соль H-18с последующим преобразованием в ортоамид H-19 (NaOMe / MeOH), и, наконец, вытеснение одной молекулы метанола путем нагревания в толуоле. Березовое сокращение H-20 (литий в жидкости аммиак, т-бутанол, THF ) при условии тетраена H-21. Обработка кислотой в тщательно контролируемых условиях сначала привела к промежуточному диона с двойной связью в положении β, γ, которая переместилась в сопряженный положение в дионе H-22, дублированный пентацикленон.[1]:528-531[14]:5 От «пентацикленона» до «корнорстерона»  Рисунок 13: Гарвардский синтез A-D-компонентов: от «пентациленона» до «корнорстерона». Этиленкеталь защитная группа в пентацикленоне H-22 был преобразован в кетоновую группу H-23 кислотно-катализируемым гидролиз.[1]:531 В диоксим в первую очередь образуется в результате реакции дикетона H-23 с гидроксиламмоний хлорид был региоселективно гидролизованный (азотистая кислота / уксусная кислота) до желаемого монооксима H-24. Это оксим стерически более затрудненный кетонная группа, атом азота которой призван стать азотом кольца D целевой молекулы. Решающим для этой цели является конфигурация у двойной связи моноксима гидроксильная группа занимает стерически менее затрудненное положение.[1]:532 Двойные связи C, C как циклопентенового, так и циклогексенонового кольца в H-24 затем были расколоты озонолиз (озон при 80 ° C в MeOH, периодная кислота ), а образовавшаяся карбоксильная группа этерифицирована CH2N2 ) на дикетон H-25. An внутримолекулярный альдольная конденсация 1,5-дикарбонильного звена в MeOH с использованием пирролидин ацетат в качестве основы, а затем тозилирование гидроксильной группы оксима, давая производное циклогексенона H-26. Второй озонолиз во влажном метилацетат с последующей обработкой периодной кислотой и CH2N2 дал H-27. Перестановка Бекмана (MeOH, сульфонат полистирола натрия, 2 часа, 170 ° C), полученный региоселективно[1]:532 лактам H-27a (не изолирован), который далее прореагировал в результате амино-карбонильной конденсации → каскад альдольной конденсации до тетрацикла H-28,[1]:533-534 называется α-коррнорстерон, считая это "краеугольным камнем"[1]:534 в синтезе желаемого A-D-компонента.[1]:531-537 Это соединение требовало сильнощелочных условий, чтобы открыть его лактам кольцо, но было обнаружено, что несовершеннолетний изомер, также выделенный из реакционной смеси, β-коррнорстерон H-29, с легкостью раскрывает это лактамное кольцо в щелочной среде.[1]:536 Структурно два изомера отличаются только ориентацией боковой цепи пропионовой кислоты в кольце A: β-изомер имеет более стабильную транс-ориентацию этой цепи по сравнению с соседней цепью уксусной кислоты, образованной после раскрытия лактамного кольца. Уравновешивание α-коррорстерона H-28 нагреванием в сильном основании с последующим подкислением и обработкой диазометан, привела к выделению чистого β-корнорстерона H-29 с доходностью 90%.[1]:537 Правильная абсолютная конфигурация шести смежных асимметричные центры в β-корнорстероне было подтверждено рентгеновский анализ кристаллической структуры бром-β-корнорстерона[69][1]:529 с "неестественной" конфигурацией.[1]:538[14]:8[4](0:49:20-0:50:42) Синтез компонента A-D, несущего функцию пропионовой кислоты в кольце D в качестве метоксикарбонильной группы (модельный компонент A-D)  Рисунок 14: Гарвардский синтез A-D-компонентов: f-недифференцированная модель A-D-компонента Лечение β-корнорстерона H-29 с метанольным HCl расщепляет лактамное кольцо и дает енольный эфир производное под названием гесперимин[примечание 15] Н-30у. Озонолиз до альдегида H-32u, восстановление альдегидной группы с NaBH4 в MeOH к первичный спирт Н-33у и, наконец, превращение гидроксильной группы через соответствующие мезилат дал бромид Н-34у. Это составляет модельный A-D-компонент, компонент с недифференцированной пропионовой кислотной функцией в кольце D (т.е. несущий группу сложного метилового эфира, как и все другие боковые цепи).[1]:539-540 Синтез A-D-компонента, несущего функцию пропионовой кислоты в кольце D в качестве нитрильной группы  Рисунок 15: Гарвардский синтез A-D-компонентов: F-дифференцированный A-D-компонент Превращение β-корнорстерона H-29 к соответствующему компоненту A-D H-34[1]:538-539 содержащую карбоксильную функцию боковой цепи пропионовой кислоты кольца D в виде нитрил группа, отличная от всех других метоксикарбонильных групп, включает следующие стадии: обработка H-29 с метанольным раствором тиофенол и HCl дает производное фенилтиоенольного эфира H-30, которые при озонолизе при низкой температуре давали соответствующие тиоэфир -альдегид H-31 и, после обработки жидким аммиаком, амид H-32. Восстановление альдегидной группы с помощью NaBH4 к H-33, мезилирование первичной гидроксильной группы с метансульфоновый ангидрид в условиях, которые также преобразуют первичную амидную группу в желаемую нитрильную группу и, наконец, замена метансульфонилоксигруппы на A-D-компонент, образующийся бромидом H-34 с функцией пропионовой кислоты в кольце D в виде нитрила, отличающейся от всех других таких боковых цепей.[1]:539-540[4](1:01:56-1:19:47) |
| Сопряжение Гарвардских A-D-компонентов с B-C-компонентом ETH |
|---|
Строительство Коррин хромофор с его тремя винилогичный амидин единиц составляет - помимо прямой одинарной связи между кольцами A и D - центральную проблему для любой попытки синтезировать витамин B12. Самый первый подход к полному синтезу витамина B12 запущен Корнфорт[43]:261-268 была прекращена, когда возникла задача связывания синтезированных кольцевых предшественников.[18]:1493,1496 Сопряжение гарвардских A-D-компонентов с B-C-компонентом ETH потребовало обширной исследовательской работы, несмотря на знания, полученные при синтезе модели ETH менее сложных (т.е. менее периферически замещенных) корринов. То, что можно назвать эпической помолвкой по формальному оформлению всего двух облигаций C, C, длилось с начала 1967 года.[18]:1557 до июня 1970 г.[2] Как в ETH, так и в Гарварде, обширные модельные исследования по объединению упрощенных энаминоид аналоги A-D-компоненты с (кольцом C) имино- и тио-иминоэфирное производное полноценного BC-компонента последовательно показали, что связывание компонентов Гарварда и ETH вряд ли может быть достигнуто с помощью метода, который оказался столь успешным в синтезе более простых корринов, а именно с помощью межмолекулярная конденсация енамино-имино (или тио-имино) сложного эфира[7][8][18]:1561[60]:41-58[1]:544[4](1:25:02-1:26:26) Результат этих модельных исследований определил окончательный тип структуры гарвардского A-D-компонента: структура, способная действовать как компонент C / D-связи посредством сульфидное сжатие посредством алкилирующего связывания,[8]:384-386[45] т.е. бромид Н-34у.[7]:18-22[60]:47,51-52 Этот метод уже был реализован группой ETH при синтезе B-C-компонент.[31]:16-19[35]:1927-1941[18]:1537-1540 Расширенный поиск оптимальных условий, сначала для C / D-связи A-D-компонента с B-C-компонентом ETH. E-19, то для условий последующего внутримолекулярного замыкания A / B-коррин-кольца в обеих лабораториях исследовали, используя f-недифференцированную модель A-D-компонента[примечание 7] Н-34у[1]:540 в качестве модели.[2]:287-300[18]:1561-1564 В результате работы Ёсито Киши в Гарварде,[2]:290[18]:1562[14]:11-12 и Питер Шнайдер из ETH,[46]:12,22-29[18]:1563-1564 optimal conditions for the C/D-coupling were eventually found at Harvard, while the first and most reliable method for the corrin-ring closure between rings A and B was developed at ETH.[18]:1562 The procedures of C/D-coupling and A/B-corrin-ring closure developed in this model series were later applied to the corresponding steps in the f-differentiated series as parts of the cobyric acid synthesis. Synthesis of dicyano-cobalt(III)-5,15-bisnor-a,b,c,d,e,f,g-heptamethyl-cobyrinate from the ring-D undifferentiated model A-D-component D/C coupling.[7]:22-23[2]:287-292[46]:12,22-28[18]:1561-1562  Figure 16: Harvard/ETH A/B approach to cobyric acid: D/C coupling of the Harvard model A-D-component with the ETH B-C-component The key problem in this step was the lability of the primary coupling product, thioether HE-35u, isomerizing to other thioethers at first not amenable to sulfide contraction in a reproducible procedure with acceptable yields.[2]:287-290[4](1:26:59-1:32:00) Induced by potassium т-butoxide in THF/т-butanol under rigorously controlled conditions with strict exclusion of air and moisture, the model A-D-component H-34u smoothly reacted with the B-C-component E-19[46]:53-58 to give the sulfur-bridged coupling product HE-35u, named "thioether type I", in essentially quantitative yield.[2]:287-288 However, this product could be isolated only under very carefully controlled conditions, since it equilibrates with extreme ease (e.g., chromatography, or traces of trifluoroacetic acid in methylenechloride solution) to the more stable isomeric thioether HE-36u (thioether type II) which contains, in contrast to thioether type I, the π-system of a conjugatively stabilized vinylogous amidine.[2]:289 Depending on conditions, still another isomer HE-37u (thiother type III) was observed.[2]:290 Starting with such mixtures of coupling products, at ETH a variety of conditions (e.g. methyl-mercury complex, BF3, triphenylphosphine[46]:58-65[2]:291) were found to induce (via HE-38u) the contraction step to HE-39u in moderate yields.[18]:1562[2]:287-292 With the choice of the solvent found to be crucial,[4](1:34:52-1:35:12) the optimal procedure at Harvard was heating thiother type II HE-36u в sulfolane in the presence of 5.3 equivalents trifluoroacetic acid and 4.5 equivalents of tris-(β-cyanoethyl)-phosphine at 60 °C for 20 hours, producing HE-39u in up to 85% yield.[2]:292[46]:65-72 Later it was discovered that nitromethane could also be used as solvent.[4](1:34:52-1:35:13)[46]:28 A/B-ring closure.[2]:293-300[46]:12,29-39[18]:1562-1564 The problem of corrin-ring closure between rings A and B was solved in two different ways, one developed at ETH, the other pursued at Harvard.[30]:19 Both methods correspond to procedures developed before in the synthesis of metal complexes[70] as well as free ligands[71] of simpler corrins.[7]:25-28[8]:387-389[18]:1563 In the explorations of ring-closure procedures for the much more highly substituted A/B-seco-corrinoid intermediate HE-39u, the ETH group focused on the intramolecular version of the oxidative sulfide contraction method, eventually leading to the dicyano-cobalt(III)-complex HE48u.[46]:29-39[2]:297-299 This first totally synthetic corrinoid intermediate was identified with a corresponding sample derived from vitamin B12.[18]:1563 At Harvard, it was shown that the closure to the corrin macrocycle could also be realized by the method of thioiminoester/enamine condensation.[2]:299-300 All reactions described here had to be executed on a very small scale, with "... the utmost rigour in the exclusion of oxygen from the reaction mixtures"[2]:296, and most of them also under strict exclusion of moisture and light, demanding very high standards of experimental expertise.[2]:304 The major obstacle in achieving an A/B-corrin-ring closure was the exposure of the highly unstable ring B exocyclic methylidene double bond, which tends to isomerize into a more stable, unreactive endocyclic position with great ease.[46]:86,97-98[2]:293-294[3]:161[18]:1562  Figure 17: Harvard/ETH A/B approach to cobyric acid: A/B-ring closure to the f-undifferentiated model 5,15-bisnorcobyrinat The problem was solved at ETH[18]:1562-1563[46]:29-39,126-135 by finding that treatment of the thiolactone-thiolactam intermediate HE-40u (obtained from HE-39u by reacting with п2S5[46]:73-83) с dimethylamine in dry MeOH (room temperature, exclusion of air and light) smoothly opens the thiolactone ring at ring B, forming by elimination of H2S the exocyclic methylidene double bond as well as a dimethylamino-amide group in the acetic acid side chain.[46]:32-34,96-99 These conditions are mild enough to prevent double bond tautomerization to the thermodynamically more stable isomeric position in the ring. Immediate conversion with a Zn-perchlorate-hexa(dimethylformamide) complex in methanol to zinc complex HE-41u, followed by oxidative coupling (0,05 mM solution of я2 /KI in MeOH, 3 h) afforded HE-42u.[46]:100-105 Sulfide contraction (triphenylphosphine, trifluoroacetic acid, 85 °C, exclusion of air and light) followed by re-complexation with Zn(ClO4)2 (KCl, MeOH, diisopropylamine ) led to the chloro-zinc complex HE-43u.[46]:105-116 The free corrinium salt formed when HE-43u was treated with trifluoroacetic acid in acetonitrile was re-complexed with anhydrous CoCl2 in THF to the dicyano-cobalt(III)-complex HE-44u.[46]:117-125[2]:295 Conversion of the dimethylamino-amide group in the acetic acid side chain of ring B into the corresponding methylester group (О-methylation by trimethyloxonium tetrafluoroborate, followed by decomposition of the iminium salt with aqueous NaHCO3) afforded totally synthetic 5,15-bisnor-heptamethyl cobyrinate HE-48u.[46]:11,117-125 A crystalline sample of HE-48u was identified via UV/VIS, ИК, и ORD spectra with a corresponding crystalline sample derived from vitamin B12[46]:42,135-141[53]:14,64-71,78-90[2]:287,301-303[3]:146-150[72] Later at Harvard,[2]:299-300 the A/B-corrin-ring closure was also achieved by converting the thiolactone-thiolactame intermediate HE-40u to thiolactone-thioiminoester HE-45u к S-methylation of the thiolactam sulfur (MeHgOi-Pr, then trimethyloxonium tetrafluoroborate). The product HE-45u was subjected to treatment with dimethylamine (as in the ETH variant), forming the highly labile methylidene derivative HE-46u, which then was converted with anhydrous CoCl2 in THF to dicyano-cobalt(III) complex HE-47u, the substrate ready to undergo the (A⇒B)-ring closure by a thioiminoester/enamine condensation. A careful search at Harvard for reaction conditions led to a procedure (KO-т-Bu, 120 °C, two weeks) that gave corrin Co complex HE-44u, identical with and in overall yields comparable with HE-44u obtained by the ETH variant of the sulfide contraction procedure.[2]:300 Since in corrin model syntheses such a C,C-condensation required induction by a strong base, its application in a substrate containing seven methylester groups was not without problems;[18]:1562 in a, milder reactions conditions were applied.[3]:162 Synthesis of dicyano-cobalt(III)-5,15-bisnor-a,b,d,e,g-pentamethyl-cobyrinate-c-N,N-dimethylamide-f-nitrile (the common corrinoid intermediate) from the ring-D-differentiated A-D-component  Figure 18: Harvard/ETH A/B approach to cobyric acid: coupling of the Harvard f-differentiated A-D-component with the ETH B-C-component to the common corrinoid intermediate The A-D-component H-34[примечание 8] with its propionic acid function at ring D differentiated from all the other carboxyl functions as nitrile group had become available at Harvard in spring 1971.[49]:23 As a result of the comprehensive exploratory work that had been done with the model A-D-component at Harvard and ETH,[2]:288-292[46]:22-28[18]:1561-1562 joining the proper A-D-component H-34 with the B-C-component E-19 by three operations H-34 + E-19 →→ HE-36 → HE-39.[3]:158-159[4](1:19:48-1:36:15) Closing the corrin ring was achieved in the sequence HE-39 (P2S5, xylene, γ-picoline )→ HE-40[4](1:36:45-1:37:49) → HE-41[4](1:37:51-1:42:33) → HE-42[4](1:42:35-1:44:34) → HE-43 (overall yield "about 60 %"[4](1:44:35-1:46:32)), and finally to cobalt complex HE-44.[4](1:46:34-1:52:51)[3]:160-166 Reactions in this sequence were based on the procedures developed in the undifferentiated model series.[2]:293-300[46]:29-39[18]:1562-1564 Two methods were available for the A/B-ring closure: oxidative sulfide contraction within a zinc complex, followed by exchange of zinc by cobalt (ETH[3]:162-165), or the Harvard alkylative variant of a sulfide contraction,[3]:160-162 thio-iminoester /enamine condensation of the cobalt complex (improved reaction conditions: diazabicyclononanone in DMF, 60 °C, several hours[3]:162). Woodward preferred the former one:[3]:165 "...the oxidative method is somewhat superior, in that it is relatively easier to reproduce, .... ".[4](1:52:37-1:53:06) The corrin complex dicyano-cobalt(III)-5,15-bisnor-pentamethyl-cobyrinate-c-N,N-dimethylamide-f-nitrile HE-44 took up the role of the common corrinoid intermediate in the two approaches to cobyric acid synthesis: HE-44 ≡ E-37. Due to the high configurational lability of C-H chirogenic centers C-3, C-8 and C-13[4](1:21:49-1:23:42,1:35:43-1:36:14,1:51:51-1:52:30) на лиганд periphery in basic or acidic milieu, separation by HPLC was indispensable for isolation, purification and characterization of pure diastereomers of this and the following corrinoid intermediates.[3]:165-166[9]:88-89[4](1:53:07-2:01:24) |
| Preparation of ring-C precursor from (+)-camphor by the Harvard group |
|---|
 Figure 19: Harvard preparation of the ring-C precursor from (+)-camphor Starting material for the synthesis of a ring-C precursor was (+)-камфорхинон H-35[note 16] which was converted to the acetoxy-trimethylcyclohexene-carboxylic acid H-36 к BF3 в acetic anhydride, a reaction pioneered by Manasse & Samuel in 1902,[73], already successfully applied in a previous synthesis of the ring-C precursor by Pelter and Cornforth.[примечание 6] Conversion of H-36 to amide H-37 was followed by its ozonolysis к peroxide H-38 which was reduced to the keto-сукцинимид H-46 by zinc and MeOH. Treatment with methanolic HCl gave lactam H-40, followed by thermal elimination of methanol to the ring-C precursor H-41[1]:540-542[46]:49-50[14]:4-5,15 This was found to be identical with the ring-C precursor E-13 prepared by a different route[примечание 5] at ETH.[59]:32[42]:30,33-34,81 |
The ETH approach to the synthesis of cobyric acid: the path to the common corrinoid intermediate via A/D-corrin-ring closure
In the A/D approach to the synthesis of cobyric acid, the four ring precursors (ring-C precursor only formally so[12]:исх. 22) derive from the two enantiomers of one common chiral starting material. All three vinylogous amidine bridges that connect the four peripheral rings were constructed by the sulfide contraction method, with the B-C-component – already prepared for the A/B-approach – serving as an intermediate.[12][11] The photochemical A/D-secocorrin→corrin cycloisomerization, by which the corrin ring was closed between rings A and D, is a novel process, targeted and found to exist in a model study (ср. fig. 2 ).[34][35]:1943-1948
| Synthesis of the ETH B-C-component (part of the A/B as well as A/D approach) |
|---|
Syntheses of the ring-B precursor Two syntheses of ring-B precursor (+)-E-5 were realized; the one starting from 2-butanone was used further.[6]:188 Two pathways for the conversion of the ring-B precursor into the ring-C precursor (+)-E-5 → (−)-E-13 ≡ H-41 were developed, one at ETH,[42]:15-39[1]:544, and one at Harvard.[6]:193[note 17] These conversions turned out to be inadequate for producing large amounts of ring-C-precursor.[44]:38[18]:1561 However, the pathway developed at ETH served the purpose of determining the absolute configuration of the ring-B precursor.[6]:193[59]:32 Bulk amounts of ring-C precursor to be used for the production of the B-C-component at ETH[42]:40[6]:193[31]:631 were prepared at Гарвард из (+)-camphor by a route originally developed by Pelter and Cornforth.[примечание 6]  Figure 20: ETH synthesis of the B-C-component: synthesis of the two enantiomers of the ring-B precursor Ring-B precursor from 2-butanone and glyoxylic acid. Aldol condensation между 2-butanone и glyoxylic acid by treatment with concentrated фосфорная кислота ) gave stereoselectively (транс)-3-methyl-4-oxo-2-pentenoic acid E-1.[37]:11-20,45-45 Diels-Alder reaction of E-1 с бутадиен in benzene in the presence of SnCl4 afforded the racemate из chiral Diels-Alder adduct E-2 который был resolved into the enantiomers by sequential salt formation with both (−)- and (+)-1-phenylethylamine.[41]:22,59-62 В chirogenic centers of the (+)-enantiomer (+)-E-2 possessed the absolute конфигурация из ring B in vitamin B12.[58]:35[6]:191 Oxidation of this (+)-enantiomer with chromic acid in acetone in the presence of sulfuric acid afforded the dilactone (+)-E-3 of the intermediary tricarboxylic acid E-3a.[41]:35,72-73 Thermodynamic control of dilactone formation leads to the cis-configuration of the ring junction.[41]:32-34 Elongation of the acetic acid side chain of (+)-E-3 посредством Arndt-Eistert reaction (via the corresponding хлорангидрид and diazoketone) gave dilactone (+)-E-4.[59]:15-16,65-67 Лечение (+)-E-4 с NH3 in MeOH at room temperature formed a dual mixture of isomeric lactam -lactones in a ratio of 2:1, with ring-B precursor (+)-E-5 predominating (isolated in 55% yield).[44]:12-17,57-63[6]:186-188[12][1]:542-543 The isomeric lactam-lactone could be isomerized to (+)-E-5 by treatment in methanolic HCl.[59]:24-26,81-84  Figure 21: ETH synthesis of the B-C-component: Alternative Synthesis of the (racemic) Ring-B Precursor (only one enantiomer shown for racemates) Alternative synthesis of racemic ring-B precursor from Hagemann's ester: implementation of the amidacetal-Claisen rearrangement. Five steps were needed to transform Hagemann's ester rac-E-6 into the racemate of the lactam-lactone rac-E-5 form of the ring-B precursor.[58]:14-31[6]:188-190 The product of the C-methylation step rac-E-6 → rac-E-7 (NaH, CH3я ) was purified via its crystalline oxime. В cis-hydroxy-ester (configuration secured by lactone formation[58]:64) resulting from the reduction step rac-E-7 → rac-E-8 (NaBH4 ) had to be separated from the транс isomer. The thermal rearrangement rac-E-8 → rac-E-9 constitutes the implementation из amidacetal-Claisen rearrangement in organic synthesis,[74][58]:36-49 a precedent to Johnson's orthoester-Claisen и Ireland's ester-enolate rearrangement.[75] Ozonolysis (О3 /MeOH, HCOOH /ЧАС2О2 ) из N,N-dimethylamide ester rac-E-9 afforded dilactone acid rac-E-10, from which two reactions led to lactam-lactone methylester rac-E-7, the racemate of ring-B precursor (+)-E-7.[58]:57-67 Determination of absolute configuration of (+)-ring-B precursor via its conversion into the (+)-ring-C precursor  Figure 22: ETH synthesis of the B-C-component: Conversion of Ring-B Precursor to Ring-C Precursor The conversion of ring-B precursor into the ring-C precursor was based on a reductive decarbonylation из thiolactone E-12 with chloro-tris-(triphenylphosphino)-rhodium(I).[42]:14-32[6]:191-193[12] Treatment of a methanolic solution of ring-B precursor (+)-E-5 with diazomethane in the presence of catalytic amounts of sodium methoxide, followed by thermal elimination of methanol, gave methylidene lactam E-11, which was converted to the thiolactone E-12 with liquid ЧАС2S containing a catalytic amount of trifluoracetic acid.[42]:15-16,56-58 Heating E-12 in toluene with the Rh(I)-complex afforded ring-C precursor (−)-E-13 besides the corresponding cyclopropane derivative E-14. Ring-C precursors prepared via this route and from (+)-camphor at Harvard [1]:540-542 were found to be identical: (−)-E-13 ≡ H-41.[42]:33-34 Ozonolysis of ring-C precursor (−)-E-13 дал сукцинимид derivative (−)-E-15.[42]:33-35,88-89 This succinimide was found to be identical[6]:193[1]:543-544 in constitution and optical rotation (i.e., configuration) with the corresponding succinimide derived from ring C of Vitamin B12, isolated after ozonolysis of crystalline heptamethyl-cobyrinate (cobester[примечание 9]) prepared from Vitamin B12.[54]:9-18,67-70 The approach pursued at Harvard for conversion of ring-B precursor into ring-C precursor was based on a photochemical degradation of the acetic acid side chain carboxyl group, starting from (+)-E-7 prepared at ETH.[note 17] Coupling of ring-B and ring-C precursors to the B-C-component. Implementation of the sulfide contraction C,C-condensation method В iminoester /enamine C,C-condensation method for constructing the vinylogous amidine system, developed in the model studies on corrin synthesis,[26][33] failed completely in attempts to create the targeted C,C-bond between ring-B precursor (+)-E-5 with ring-C precursor (−)-E-13 to give the B-C-component E-18.[6]:193-194[8]:379[1]:544 The problem was solved by "intramolecularization" of the bond formation process between the electrophilic (thio)iminoester carbon and the nucleophilic methylidene carbon of the enamine system through first oxidatively connecting these two centers by a sulfur bridge, and then achieving the C,C-bond formation by a now intramolecular thio-iminoester/enamine condensation with concomitant transfer of the sulfur to a thiophile.[6]:194-197[8]:380-386[18]:1537-1538 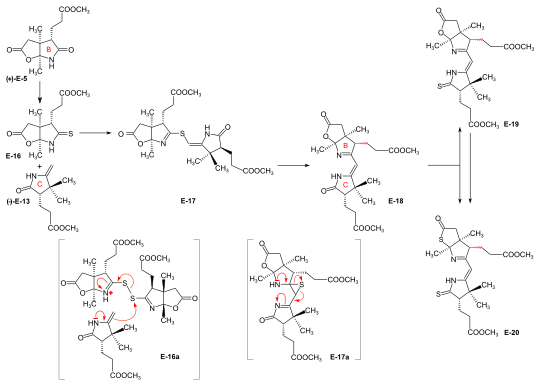 Figure 23: ETH synthesis of the B-C-component: coupling of the ring B and C precursors (implementation of C/C-coupling by the sulfide-contraction method) Conversion of lactam (+)-E-5 into the corresponding thiolactam E-16 (P2S5),[44]:20-23,74-75 oxidation of E-16 с перекись бензоила in the presence of ring-C precursor (−)-E-13 (prepared at Harvard by the Cornforth route[примечание 6]), followed by heating the reaction product E-17 в triethylphosphite (as both solvent and thiophile) afforded B-C-component E-18 as a (not separated) mixture of two epimers (regarding the configuration of the propionic side chain at ring B) in up to 80 % yield.[44]:38-43,96-102[31]:16-19[8]:381-383[46]:20-21,50-52 The bracketed formulae in the reaction scheme illustrate the type of механизм operating in the process: E-16a = primary coupling of E-12 и E-10 к E-13; E-17a = extrusion of the sulfur atom (captured by thiophile) to E-14, where it is left open whether this latter process occurs at the stage of the episulfide. This reaction concept developed at this stage, dubbed sulfide contraction,[6]:199[45][18]:1534-1541[35]:1927-1941 turned out to make possible the construction of all three meso-carbon bridges of the vitamin's corrin ligand in both approaches of the synthesis.[12][11][2]:288-292,297-300[3]:158-164 The conversion of bicyclic lactone-lactam E-18 into the corresponding thiolactone-thiolactam E-20 was brought about by heating with п2S5 /4-methylpyridine в xylene at 130 °C; milder condition produced thiolactam-lactone E-19, used for coupling with the Harvard A-D-components.[49]:73-83 |
| Coupling of the B-C-component with ring-D and ring-A precursors |
|---|
Synthesis of ring-D precursor for the A/D approach 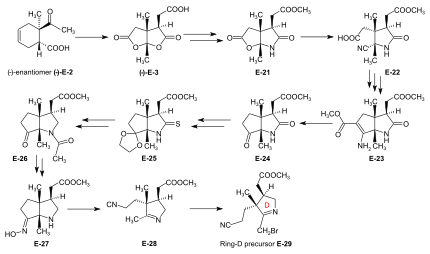 Figure 24: ETH A/D approach to cobyric acid: synthesis of ring-D precursor The starting material for the ring-D precursor,[59]:40-61[61]:17-22[12] the (−)-enantiomer of the dilactone-carboxylic acid (−)-E-3, was prepared from the (−)-enantiomer of the Diels-Alder adduct (−)-E-2[note 18] by oxydation with chromic acid /sulfuric acid in acetone.[41]:35,72-73 Лечение (−)-E-3 with NH3 in MeOH gave a lactone-lactam-acid which was esterified with diazomethane to the ester E-21,[59]:104-110 the lactone ring of which was opened with KCN in MeOH to give E-22.[59]:114-116 Conventional conditions of an Arndt-Eistert reaction (SOCl2: acid chloride, then CH2N2 in THF: diazoketone, treated with Ag2О in MeOH) led to an – unforeseen, yet useful – ring closure of the originally formed chain-elongated ester through participation of the cyano group as a neighboring electrophile, affording the bicyclic enamino-ester derivative E-23.[59]:116-120 Hydrolysis with aqueous HCl, accompanied by decarboxylation, and re-esterification with diazomethane gave keto-lactam-ester E-24.[59]:123-126[61]:40-41 Ketalization ((CH2OH)2, CH(OCH3)3, TsOH ) из E-24 and conversion of this lactam-ester to thiolactam E-25 (п2S5 ) was followed by reductive removal of the sulfur with Raney nickel, acetylation of the amino group, and hydrolysis of the ketal (AcOH) to afford E-26.[61]:42-59 This was converted by deacetylation of the amino group with HCl, and then by treatment with NH2OH/HCl, MeOH/NaOAc into oxime E-27. Beckmann fragmentation (HCl, SOCl2 in CHCl3, N-polystyryl-piperidine) of this oxime E-27 produced imino-nitrile E-28,[61]:60-67 which, when treated with бром (in MeOH, phosphate buffer pH 7.5, -10 °C) gave ring-D precursor E-29.[49]:84-88 Conversion of the ring-B precursor into the ring-A precursor for the A/D approach  Figure 25: ETH A/D approach to cobyric acid: conversion of ring-B precursor into ring-A precursor The ring-A precursor (−)-E-31 required in the A/D approach is a close derivative of ring-B precursor (+)-E-5. Its preparation from (+)-E-5 required opening of the lactone group (KCN in MeOH), followed by re-esterification with diazomethane to E-30, then conversion of the lactam group into a thiolactam group with P2S5 to yield (−)-E-31.[49]:63-72[12] Coupling of the B-C-component with ring-D and ring-A precursors The most efficient way of attaching the two rings D and A to the B-C-component E-18 was to convert E-18 directly into its thiolactam-thiolactone derivative E-20 and then to proceed by first coupling ring-D precursor E-29 to ring C, and then ring-A precursor E-31 to ring B, both by the sulfide contraction method.[49]:26-31[9]:80-83[12] The search for the reaction conditions for these attachments was greatly facilitated by exploratory work done on the two sulfide contraction steps in the A/B approach model study.[49]:27[46]:22-39[2]:285-300  Figure 26: ETH A/D approach to cobyric acid: Attaching ring-C and ring-A precursors to the B-C-component to yield the A/D-seco-corrin Attachment of ring-D precursor E-29 to the ring-C thiolactam in E-20 by sulfide contraction via alkylative coupling (т-BuOK in т-BuOH/THF, tris-(β-cyano-ethyl)-phosphin/CF3COOH в sulfolane ) afforded the B/C/D-sesqui-corrinoid E-32.[49]:89-97 To attach ring-A precursor E-31, the ring B of E-32 was induced to expose its exocyclic methylidene double bond by treatment with dimethylamine in MeOH (using the method[note 19] developed by Schneider[46]:32-34) forming E-33[49]:108-115 which was subjected to the following cascade of operations:[49]:130-150 iodination (N-iodosuccinimide, CH2Cl2, 0°), coupling with the thiolactam sulfur of the ring-A precursor E-31 [(CH3)3Si]2N-Na in benzene/т-BuOH), complexation (Cd(ClO4)2 in MeOH), treatment with triphenylphosphine /CF3COOH in boiling benzene (sulfide contraction) and, finally, re-complexation with Cd(ClO4)2/N,N-diisopropylethylamine in benzene/MeOH). These six operations, all carried out without isolation of intermediates, gave A/D-seco-corrin complex E-34 as mixture of peripheral epimers (separable via HPLC[49]:143-147) in 42-46 % overall yield.[49]:139 |
| A/D-corrin-ring closure by the photochemical A/D-seco-corrin→corrin cycloisomerization |
|---|
A/D-corrin-ring closure by the photochemical A/D-seco-corrin→corrin cycloisomerization to dicyano-cobalt(III)-5,15-bisnor-a,b,d,e,g-pentamethyl-cobyrinate-c-N,N-dimethylamide-f-nitrile (the common corrinoid intermediate) The conditions and prerequisites for the final (A⇒D)-corrin -ring closure were taken over from extensive corrin model studies.[34][76][9]:71-74,83-84[18]:1565-1566[35]:1942-1962 Problems specific to the cobyric acid synthesis that had to be tackled were:[9]:84-88 the possible formation of two diastereomeric A/D-транс-junctions in the ring closure,[49]:37-38 exposure of the methylidene double bond at ring A of the A/D-seco-corrin E-34 in a labile Cd complex,[49]:35-36[18]:1566 and epimerizability of the peripheral stereogenic centers C-3, C-8 and C-13 before and after ring closure.[49]:39[3]:148-150 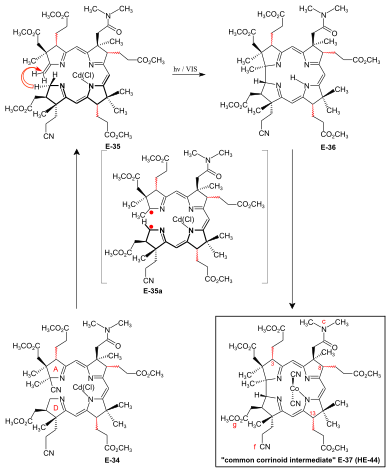 Figure 27: ETH A/D approach to cobyric acid: photochemical A/D-seco-corrin→corrin cycloisomerization to the common corrinoid intermediate In the application of this novel process in the A/D approach of the cobyric acid synthesis,[9]:86-95[49]:39-53[12]:1419 the reaction proceeded most efficiently and with highest coil stereoselectivity in favor of the natural A/D-транс junction in an A/D-seco-corrin cadmium complex.[49]:42-45[3]:166 Treatment of Cd-complex E-34 as mixture of peripheral epimers с 1,8-Diazabicyclo(5.4.0)undec-7-ene в sulfolane at 60 °C under strict protection against light to eliminate the cyano group at ring A, directly followed by re-treatment with Cd(ClO4)2, led to labile[49]:172 A/D-seco-corrin complex E-35 as a mixture of peripheral epimers. This was directly subjected to the key step, the photochemical ring closure reaction under rigorous exclusion of air:[49]:40 visible light, under Аргон, MeOH, AcOH, 60° C. Product of the A/D-ring closure was the free corrin лиганд E-36, as the originally formed Cd-corrinate – in contrast to the Cd-seco-corrinate E-35 – decomplexes in the reaction medium.[49]:173[12]:1419 Corrin E-36 was immediately complexed (CoCl2,[18]:1499-1500,1563-64 KCN, air, H2O, CH2Cl2) and finally isolated (thick-layer chromatography ) as mixture of peripheral epimers in 45-50 % yield over four operations:[49]:169-179 the common corrinoid intermediate dicyano-cobalt(III)-complex E-37 ≡ HE-44.[note 20] HPLC analysis of this mixture E-37 showed the presence of six epimers with natural ligand helicity (Σ 95%, CD spectra ), among them 26% of natural diastereomer 3α,8α,13α, and an equal amount of its C-13 нео-epimer 3α,8α,13β.[49]:46,179-186[12]:1414 Two HPLC fractions (Σ 5%) contained diastereomers with unnatural ligand helicity, as shown by inverse CD spectra.[49]:42-43 Product mixtures from several such cycloisomerizations were combined for preparative HPLC separation and full characterization of the 14 isolated diastereomers of E-37[49]:207-251 (of 16 theoretically possible, regarding helicity and the epimeric centers C-3, C-8, C-13[49]:39).  Figure 28: ETH A/D approach to cobyric acid: coil selectivity in A/D-ring closure In an analytical run, the mixture of cadmium-seco-complex epimers E-35 was separated by HPLC (in the dark) into the natural chloro-cadmium-3α,8α,13α-A/D-seco-corrinate diastereomer (ααα)-E-35 and four other epimer fractions[49]:281-293 Upon irradiation[49]:53[12] and following cobaltation, (ααα)-E-35 произведено E-37 in yields of 70-80% as an essentially dual mixture of mainly the 3α,8α,13α epimer, besides some 3α,8α,13β epimer. Less than 1% of fractions with unnatural coil were formed (HPLC, UV/VIS, CD ).[49]:293-300 Mechanistically, то photochemical A/D-seco-corrin corrin cycloisomerization involves an antarafacial sigmatropic shift of the α-hydrogen of the CH2 position C-19 at ring D to the CH2 position of the methylidene group at ring A within a triplet excited state, creating a transient 15-center-16-electron π-system (see E-35a в fig. 27 ) that antarafacially collapses between positions C-1 and C-19 to the corrin system.[34][35]:1946,1967-1993[77] The coil selectivity of the ring closure in favor of the corrin ligand's natural helicity is interpreted as relating to the difference in steric hindrance between the g-methoxycarbonyl acetic acid chain at ring D and the methylidene region of ring A in the two possible helical coil configurations of the A/D-seco-corrin complex (fig. 28).[49]:38[35]:1960-1962 |
ETH/Harvard: the jointly executed final steps from the common corrinoid intermediate to cobyric acid
The final steps from the common corrinoid intermediate E-37/HE-44 to cobyric acid E-44/HE-51 were carried out by the two groups collaboratively and in parallel, the ETH group working with material produced by the A/D approach, а Гарвард group with that from the A/B approach.[61]:15[53]:22[55]:47[14]:12[18]:1570-1571 What the two groups in fact accomplished thus were the common final steps of two different syntheses.[11][12]
The tasks in this end phase of the project were the regioselective introduction of methyl groups at the two meso positions C-5 and C-15 of E-37/HE-44, followed by conversion of all its peripheral carboxyl functions в primary amide groups, excepting that in side chain f at ring D, which had to end up as free carboxyl. These conceptually simple finishing steps turned out to be rather complex in execution, including unforeseen pitfalls like a dramatic loss of precious synthetic material in the so-called "Black Friday" (July 9, 1971).[53]:39-40,107-118[9]:97-99[3]:168-169[5](0:07:54-0:09:33)[18]:1568-1569
| Introduction of methyl groups in two meso positions |
|---|
 Figure 29: ETH/Harvard joint final steps: Introduction of methyl groups at the meso positions C-5 and C-15 This introduction of methyl groups could draw on exploratory studies on model corrins[7]:13-14[8]:375-377[78][18]:1528,1530-1532 as well as on exploratory experiments carried out at ETH on cobester[примечание 9] and its (c→C-8)-lactone derivative.[53]:27-43 Chloromethyl benzyl ether alkylated the meso position C-10 of cobester, but not that of the corresponding lactone, the difference in behavior reflecting the difference in steric hindrance exerted on the meso position C-10 by its neighboring substituents.[53]:37-39 This finding was decisive for the choice of the substrate to be used for introducing methyl groups at meso positions C-5 and C-10 of E-37/HE-44.[9]:96-99[53]:19[3]:167[18]:1567-1568 In this final phase of the synthesis, HPLC again turned out to be absolutely indispensable for separation, isolation, characterization and, above all, identification of pure isomers of dicyano-cobalt(III)-complexes of totally as well as partially synthetic origin.[9]:96-102[3]:165[53]:61-63[5](0:21:13-0:25:28)[18]:1566-1567 The first step was to convert the c-N,N-dimethylcarboxamide group of E-37/HE-44 into the (c→C-8)-lactone derivative E-38/HE-45 by treatment with йод /AcOH effecting iodination at C-8, followed by intramolecular О-alkylation of the carboxamide group to an iminium salt that hydrolyses to the lactone.[61]:23,90-108[3]:166-167[4](2:02:18-2:09:02) This lactonization leads to cis-fused rings.[53]:19[5](0:09:34-0:10:43) Reaction of (c→C-8)-lactone E-38/HE-45 with chloromethyl benzyl ether in acetonitrile in the presence of LiCl gave, besides mono-adduct, the bis-benzyloxy adduct E-39/HE-46. When treated with thiophenol, this produced the bis-phenylthio-derivative E-40/HE-47. Treatment with Raney nickel in MeOH not only set free the two methyl groups at the meso positions, but also reductively opened the lactone ring to the free c-carboxyl group at ring B, producing the correct α-конфигурация at C-8. Esterification of c-carboxyl with diazomethane afforded hexamethylester-f-nitrile E-41/HE-48.[53]:19-21,39-43,146-205[3]:167-169 For steric reasons, only the predominant[53]:19[61]:24[4](2:08:20-2:09:02) C-3 α-epimer (with the C-3 side chain below the plane of the corrin ring) reacted to a 5,15-disubstituted товар E-38/H-45, the reaction thus amounting to a chemical separation of the C-3 epimers.[53]:40[5](0:12:51-0:14:33,0:15:56-0:16:24) In improved procedures developed at Harvard later in 1972,[18]:1569 footnote 62 the reagent chloromethyl benzyl ether was replaced by формальдегид /sulfolane/HCl in acetonitrile for the alkylation step, and Raney nickel in the reduction step was replaced by zinc/acetic acid to give E-41/HE-48.[5](0:00:32-0:21:12) |
| Dicyano-cobalt(III)-3α,8α,13α-a,b,c,d,e,g-hexamethyl-cobyrinate-f-amide: Identification with material derived from vitamin B12 |
|---|
Figure 30: ETH/Harvard joint final steps: hexamethylcobyrinate-f-amide (synthesis and identification) to cobyric acid Concentrated H2ТАК4 at room temperature converted the nitrile function of pure (3α,8α,13α)-E-41/HE-48 into the primary f-amide группа E-42/HE-49, besides partial epimerization at C-13;[9]:100-103[53]:21,134-136[3]:150-151,169-170 an alternative procedure for the selective f-nitrile→f-amide conversion (BF3 in CH3COOH) later developed at Harvard proceeded without эпимеризация at C-13.[18]:1569 footnote 62[5](0:46:40-0:49:45)[53]:21 A crystalline sample of the 3α,8α,13α-epimer of dicyano-cobalt (III)-a,b,c,d,e,g-hexamethyl-cobyrinate-f-amide E-42/HE-49, isolated by HPLC, was the first totally synthetic intermediate to be chromatographically and spectroscopically identified with a relay sample made from vitamin B12.[53]:136-141[3]:170 In the remaining steps of the synthesis, only epimerization at C-13 played an important role,[53]:19-21 with 13α being the configuration of the natural corrinoids, and 13β known as нео-epimers of vitamin B12 and its derivatives;[3]:169-170[79] these are readily separable by HPLC.[5](0:19:30-0:20:21)[53]:135,208-209 In the course of 1972, comprehensive identifications (HPLC, UV/VIS, ИК, NMR, CD, mass spectra ) of crystalline samples of totally synthetic intermediates with the corresponding compounds derived from vitamin B12 were carried out in both laboratories: individually compared and identified were the 3α,8α,13α and 3α,8α,13β нео-epimer of f-amide E-42/HE-49, as well as the corresponding pair of C-13-epimeric nitriles E-41/HE-48.[53]:206-221[55]:46-47[5](0:27:28-0:46:32) All these dicyano-cobalt(III)-complexes are soluble in organic solvents[54]:11 in which the separation power of HPLC by far exceeds that of analytical methods operating in water,[53]:44-45 the solvent in which cobyric acid was to be identified, and where it exists as two easily equilibrating aquo-cyano complexes, epimeric regarding the position of the two non-identical axial Co лиганды.[61]:196-197[55]:49-60 These thorough identifications of the totally synthetic with partially synthetic materials mark the accomplishment of the two syntheses. They also reciprocally provided structure proof for a specific constitutional isomer isolated from a mixture of isomeric mono-amides formed in the partial ammonolysis of the B12-derived cobester,[примечание 9] tentatively assigned to be the 3α,8α,13α-f-amide E-42/HE-49 (see fig. 30).[54]:9-18,67-70[53]:226-239[57] |
| Synthetic cobyric acid |
|---|
The final task of reaching cobyric acid from f-amide E-42/HE-49 required the critical step of hydrolysing the singular amide function into a free carboxyl function without touching any of the six methoxycarbonyl groups around the molecule's periphery. Since exploratory attempts by the conventional method of amide hydrolysis via nitrosation led to detrimental side reactions at the хромофор, a novel way of "hydrolysing " the f-amide group without touching the six methylester groups was conceived and explored at ETH: treatment of f-amide E-42/HE-49 (B12-derived relay material) with the unusual reagent α-chloro-propyl-(N-cyclohexyl)-nitrone[80] и AgBF4 in CH2Cl2, then with HCl in H2O/dioxane, and finally with dimethylamine in isopropanol afforded the f-acid E-43/HE-50 in 57% yield.[61]:24-25,159-172[3]:170-172[5](0:53:17-0:58:30) Sustained experimentations at Harvard eventually showed the nitrosation method to be successful (N2О4, CCl4, NaOAc ) and to produce the f-carboxyl group even more effectively.[3]:172-173[5](0:58:19-0:59:15) It was also at Harvard that conditions for the last step were explored, conversion of all remaining ester groups into primary amide groups by ammonolysis. Жидкость ammonia в ethylene glycol, in the presence of NH4Cl and the absence of oxygen, converted f-carboxy-hexamethylester E-43/HE-50 into f-carboxy-hexa-amide E-44/HE-51 (= cobyric acid).[3]:173-175[53]:24 This was crystallised and shown both as the α-cyano-β-aquo and the α-aquo-β-cyano form to be chromatographically and spectroscopically identical with the corresponding forms of natural cobyric acid.[5](0:59:53-1:09:58)[3]:175-176[61]:26-27,196-221 At Harvard, the transformation E-43/HE-50 → E-44/HE-51 was eventually carried out starting with f-amide that had been obtained by total synthesis via the A/B approach.[55]:47-61 The ETH group contented itself with a corresponding f-amide → cobyric acid conversion and subsequent cobyric acid identification where the actual starting material f-amide was derived from vitamin B12.[53]:22[61]:15[12]:footnote 45[18]:1570-1571 |
Примечания
- ^ For a review about syntheses of corrins, see[25]; this includes more recent synthetic approaches to vitamin B12 by the groups of Stevens,[25]:293-298 Jacobi,[25]:298-300 и Mulzer,[25]:300-301 as well as references to approaches by Тодд или же Cornforth (see also[43]:261-268) preceding the efforts by Eschenmoser и Woodward.[18]:1493-1496
- ^ а б c d е Formulae in figs. 4 и 6 illustrate the atom, ring, and side chain enumeration in corrins: "Nomenclature of Corrinoids". Pure and Applied Chemistry. 48 (4): 495–502. 1976. Дои:10.1351/pac197648040495.
- ^ The year 1964 refers to the first corrin synthesis of a pentamethylcorrin via A/B-cyclization by iminoester/enamine-C,C-condensation;[26] то heptamethylcorrin shown here (M = Co(CN)2) was prepared by the same ring closure method in 1967.[27]
- ^ а б Friedrich, W.; Gross, G.; Bernhauer, K.; Zeller, P. (1960). "Synthesen auf dem Vitamin-B12-Gebiet. 4. Mitteilung. Partialsynthese von Vitamin B12". Helvetica Chimica Acta. 43 (3): 704–712. Дои:10.1002/hlca.19600430314. For recent partial syntheses of vitamin B12 и coenzyme B12 from cobyric acid, see Widner, Florian J.; Gstrein, Fabian; Kräutler, Bernhard (2017). "Partial Synthesis of Coenzyme B12 from Cobyric Acid". Helvetica Chimica Acta. 100 (9): e1700170. Дои:10.1002/hlca.201700170.
- ^ а б Видеть Determination of absolute configuration of (+)-ring-B precursor via its conversion into the (+)-ring-C precursor in (Show/Hide) "Synthesis of the ETH B-C-component (part of the A/B as well as A/D approach) ".
- ^ а б c d Письмо от J. W. Cornforth to A. Eschenmoser, April 16th, 1984, see [18]:1561 footnote 51; see also refs.[6][42]:40[43]:265. This preparation of a ring-C precursor from (+)-camphor involved 8 steps, compared to 4 steps[примечание 5] from the ETH ring-B precursor (but it used a commonly available precursor instead of "precious" material!)
- ^ а б Видеть Synthesis of the A-D-component carrying the propionic acid function at ring D as methoxycarbonyl group (model A-D-component) in (Show/Hide) "The Harvard synthesis of the A-D-components for the A/B approach ".
- ^ а б Видеть Synthesis of the A-D-component carrying the propionic acid function at ring D as nitrile group in (Show/Hide) "The Harvard synthesis of the A-D-components for the A/B approach ".
- ^ а б c d е Cobester (dicyano-Co-cobyrinic acid heptamethylester) is a non-natural cobyric acid derivative that had played an important subsidiary role in the B12 total syntheses;[53]:14,21,51–90,222–260 it is prepared in one step from vitamin B12 by acid-catalyzed methanolysis.[54]:9–18
- ^ "University of Bristol. WILSON BAKER SYMPOSIUM: Previous Wilson Baker lectures" (PDF). Получено 2019-10-29.. See also Eschenmoser lecture announcements in "Notizen". Nachrichten aus Chemie und Technik. 20 (5): 89–90. 1972. Дои:10.1002/nadc.19720200502..
- ^ а б c Research reports of the Harvard postdoctoral fellows involved in the vitamin B12 synthesis are in the Harvard archives; видеть "Collection: Papers of Robert Burns Woodward, 1873-1980, 1930-1979 | HOLLIS for Archival Discovery". Получено 2019-10-29..
- ^ The only "joint publication" is a 1972 interview with Eschenmoser and Woodward in Basle; [29] смотрите также[18]:1572–1574[62]:1478.
- ^ References given here are a selection from about 50 publications where these epochal syntheses are discussed in more or less detail. Они также используются для обучения синтезу натуральных продуктов на продвинутых курсах или семинарах исследовательских групп, например, Эшенмозер, А. (2001). «Эпилог: Синтез коэнзима B12: Средство обучения органическому синтезу ». In Quinkert, Gerhard; Kisakürek, M. Volkan (eds.). Очерки современной химии: от структуры молекулы к биологии. Цюрих: Verlag Helvetica Chimica Acta. С. 391–441. Дои:10.1002 / 9783906390451.ch12. ISBN 9783906390284..
- ^ Это единственная часть статей из Гарварда, опубликованных на данный момент с полными экспериментальными деталями: Флеминг, Ян; Вудворд, Р. Б. (1973). «Синтез (-) - (R) -транс-β- (1,2,3-триметилциклопент-2-енил) акриловой кислоты». Журнал химического общества, Perkin Transactions 1: 1653–1657. Дои:10.1039 / P19730001653.Флеминг, Ян; Вудворд, Р. Б. (1968). «Экзо-2-гидроксиэпикамфора». Журнал химического общества C: Органический: 1289. Дои:10.1039 / J39680001289..
- ^ Это название левой части («западная половина») строительного блока относится к Геспериды, то Нимфы Запада, как и Гесперидиум и (химически совершенно не связанные) Гесперидин;[1] ср. другие красочные наименования Вудворда: пентацикленон,[1]:530 коррнорстерон;[1]:534 корригенолид, корригенат: Коррин-генeating seco-corrins.[2]:285,296 Группа ETH назвала свой правый строительный блок «(тио) декстролином» на основе «dexter», латинского «право».[1]:538-539
- ^ Камфорхинон получают из камфоры путем реакции с диоксид селена: видеть Уайт, Джеймс Д .; Wardrop, Duncan J .; Сандерманн, Курт Ф. (2002). Проверено Кенджи Кога, Кей Манабе, Кристофером Э. Нейппом и Стивеном Ф. Мартином. «Камфорхинон и моноксим камфорхинона». Органический синтез. 79: 125. Дои:10.15227 / orgsyn.079.0125..
- ^ а б Вик, Александр: Часть I отчета, Гарвардский университет 1967 (неопубликованный[примечание 11]), цитируется в[42]:38–39.
- ^ Видеть Синтезы предшественника кольца B в (Показать / Скрыть) "Синтез B-C-компонента ETH ".
- ^ Видеть Застежка-кольцо A / B в (Показать / Скрыть) "Сопряжение Гарвардских A-D-компонентов с B-C-компонентом ETH ".
- ^ Видеть Синтез дицианокобальта (III) -5,15-биснор-a, b, d, e, g-пентаметилкобиринат-c-N, N-диметиламид-f-нитрил (обычное промежуточное соединение корриноидов) из дифференцированного по кольцу D A-D-компонента в (Показать / Скрыть) "Сопряжение Гарвардских A-D-компонентов с B-C-компонентом ETH ".
Рекомендации
- ^ а б c d е ж грамм час я j k л м п о п q р s т ты v ш Икс у z аа ab ac объявление ае аф аг ах ай эй ак аль являюсь ан Вудворд, Р. Б. (1968). «Последние достижения химии натуральных продуктов». Чистая и прикладная химия. 17 (3–4): 519–547. Дои:10.1351 / pac196817030519.
- ^ а б c d е ж грамм час я j k л м п о п q р s т ты v ш Икс у z аа ab ac объявление ае аф аг ах ай Вудворд, Р. Б. (1971). «Последние достижения химии натуральных продуктов». Чистая и прикладная химия. 25: 283–304. Дои:10.1351 / pac197125010283.
- ^ а б c d е ж грамм час я j k л м п о п q р s т ты v ш Икс у z аа ab ac объявление ае аф аг ах ай эй ак аль Вудворд, Р. Б. (1973). «Общий синтез витамина B12". Чистая и прикладная химия. 33: 145–178. Дои:10.1351 / pac197333010145. PMID 4684454.
- ^ а б c d е ж грамм час я j k л м п о п q р s т ты v ш Вудворд, Роберт Б. (27 ноября 1972 г.). Лекция Р. Б. Вудворда о полном синтезе витамина B12 - Часть 1 (записанная лекция). Введение Дэвида Долфина. Гарвардский университет, Кембридж, Массачусетс (США): YouTube. Получено 2020-01-25.
- ^ а б c d е ж грамм час я j k л м п о Вудворд, Роберт Б. (27 ноября 1972 г.). Лекция Р. Б. Вудворда по общему синтезу витамина B12 - Часть 2 (записанная лекция). Гарвардский университет, Кембридж, Массачусетс (США): YouTube. Получено 2020-01-25.
- ^ а б c d е ж грамм час я j k л м п о п Эшенмозер, А. (1968). "Die Synthese von Corrinen". Moderni Sviluppi della Sintesi Organica (X Corso estivo di chimica, Fondazione Donegani, Frascati 25.9.-5.10.1967) (на немецком). Рома: Accademia Nazionale dei Lincei. С. 181–214. ISBN 8821804054. ISSN 0515-2216.
- ^ а б c d е ж грамм час я Эшенмозер, А. (1968). «Современные аспекты синтеза корриноидов». Труды конференции Фонда Роберта А. Уэлча по химическим исследованиям. 12: 9–47. ISSN 0557-1588.
- ^ а б c d е ж грамм час я j k л м п о Эшенмозер, А. (1970). «Столетняя лекция (прочитана в ноябре 1969 г.). Дороги к корринам». Ежеквартальные обзоры, Химическое общество. 24 (3): 366–415. Дои:10,1039 / qr9702400366.
- ^ а б c d е ж грамм час я j k л м п Эшенмозер, А. (1971). Исследования по органическому синтезу. XXIII Международный конгресс чистой и прикладной химии: специальные лекции, прочитанные в Бостоне, США, 26-30 июля 1971 г. 2. Лондон: Баттервортс. С. 69–106. Дои:10.3929 / ethz-a-010165162. HDL:20.500.11850/84699. ISBN 0-408-70316-4.
- ^ а б c d е ж Fuhrer, W .; Schneider, P .; Schilling, W .; Wild, H .; Schreiber, J .; Эшенмозер, А. (1972). "Totalsynthese von Vitamin B12: die photochemische Secocorrin-Corrin-Cycloisomerisierung ". Chimia (конспект лекции). 26: 320.Maag, H .; Obata, N .; Холмс, А .; Schneider, P .; Schilling, W .; Schreiber, J .; Эшенмозер, А. (1972). "Totalsynthese von Vitamin B12: Endstufen ". Chimia (конспект лекции). 26: 320.
- ^ а б c d е ж грамм час я j k л Эшенмозер, А. (1974). "Organische Naturstoffsynthese heute. Витамин B12 как Beispiel ". Die Naturwissenschaften. 61 (12): 513–525. Bibcode:1974NW ..... 61..513E. Дои:10.1007 / BF00606511. PMID 4453344.
- ^ а б c d е ж грамм час я j k л м п о п q р s т ты v ш Икс Эшенмозер, А.; Винтнер, К. (1977). «Синтез натуральных продуктов и витамин B12". Наука. 196 (4297): 1410–1420. Bibcode:1977Научный ... 196.1410E. Дои:10.1126 / science.867037. PMID 867037.
- ^ а б c d е ж Засс, Э. (2014). "Замечательного общего синтеза, еще не опубликованного в экспериментальных деталях - витамин B12 (Слайды лекции о вручении премии Сколника на 248-м Национальном собрании ACS, Сан-Франциско, Калифорния, 12 августа 2014 г.) ». SlideShare. LinkedIn. Получено 2020-01-25. Смотрите также Уорр, Венди (2014). "Симпозиум премии Германа Сколника в честь Энгельберта Засса". Бюллетень химической информации. 66 (4 / зима 2014 г.): 37–40. Получено 2020-01-25.
- ^ а б c d е ж грамм час Крейг, Дж. Уэйн (2016). «Полный синтез витамина B12 - братство ринга ». Журнал порфиринов и фталоцианинов. 20: 1–20. Дои:10.1142 / S1088424615500960.
- ^ а б Николау, К.С.; Соренсен, Э. Дж. (1996). "Глава 8: Витамин B12. Р. Б. Вудворд и А. Эшенмозер (1973) ". Классика в полном синтезе: цели, стратегии, методы. Weinheim: VCH Verlag Chemie. стр.99 -136. ISBN 978-3-527-29231-8.
- ^ Марко, И. Э. (2001). «Синтез натуральных продуктов: искусство полного синтеза». Наука. 294 (5548): 1842–1843. Дои:10.1126 / science.1067545. PMID 11729290.
- ^ а б c d е ж грамм час я j k л м п Эшенмозер, А. (2001). RBW, витамин B12и сотрудничество Гарварда и ETH ". В Бенфей, О. Теодор; Моррис, Питер Дж. Т. (ред.). Роберт Бернс Вудворд - архитектор и художник в мире молекул. Серия «История современных химических наук». Филадельфия: Фонд химического наследия. С. 23–38. ISBN 978-0941901253. ISSN 1069-2452.
- ^ а б c d е ж грамм час я j k л м п о п q р s т ты v ш Икс у z аа ab ac объявление ае аф аг ах ай эй ак аль являюсь ан ао ap водный ар в качестве в au средний ау топор ай az ба bb до н.э bd быть парень bg бх би Ъ bk бл Эшенмозер, Альберт (2015). "Corrin Syntheses. Часть I". Helvetica Chimica Acta. 98 (11–12): 1483–1600. Дои:10.1002 / hlca.201400277.
- ^ а б Николау, К.С.; Соренсен, Э. Дж .; Винссингер, Н. (1998). "Искусство и наука синтеза органических и натуральных продуктов". Журнал химического образования. 75 (10): 1225–1258. Bibcode:1998JChEd..75.1225N. Дои:10.1021 / ed075p1225.
- ^ Николау, К.С.; Вурлумис, Дионисий; Винссингер, Николас; Баран, Фил С. (2000). «Искусство и наука полного синтеза на заре двадцать первого века». Angewandte Chemie International Edition. 39 (1): 44–122. Дои:10.1002 / (SICI) 1521-3773 (20000103) 39: 1 <44 :: AID-ANIE44> 3.0.CO; 2-L. PMID 10649349.
- ^ Эшенмозер, Альберт (1988). "Витамин B12: Эксперименты относительно происхождения его молекулярной структуры ». Angewandte Chemie International Edition на английском языке. 27: 5–39. Дои:10.1002 / anie.198800051.
- ^ Ходжкин, Дороти Кроуфут; Кампер, Дженнифер; Маккей, Морин; Пикворт, Дженни; Трублад, Кеннет Н.; Уайт, Джон Г. (1956). «Структура витамина B12". Природа. 178 (4524): 64–66. Bibcode:1956Натура.178 ... 64H. Дои:10.1038 / 178064a0. PMID 13348621.
- ^ Glusker, Дженни П. (1995). "Витамин B12 и B12 Коэнзимы ». Витамины и гормоны. 50: 1–76.
- ^ а б Bernhauer, K .; Dellweg, H .; Фридрих, В .; Гросс, Гизела; Вагнер, Ф. (1960). "Notizen: витамин B12-Faktor V1a, ein neuer "inkompletter" Grundkörper der Vitamin B12-Группа ». Zeitschrift für Naturforschung B. 15 (5): 336–337. Дои:10.1515 / znb-1960-0522.
- ^ а б c d е ж Монфортс, Франц-Петер; Осмерс, Мартина; Леупольд, Деннис (2012). «Химический синтез искусственных корринов». В Кадиш, Карл М .; Смит, Кевин М .; Гилард, Роджер (ред.). Справочник по порфириновым наукам. 25. Мировое научное издательство. С. 265–307. Дои:10.1142/9789814397605_0020. ISBN 978-981-4397-66-7.
- ^ а б c d Bertele, E .; Boos, H .; Дуниц, Дж. Д.; Elsinger, F .; Эшенмозер, А.; Felner, I .; Gribi, H.P .; Gschwend, H .; Meyer, E. F .; Pesaro, M .; Шеффолд Р. (1964). «Синтетический путь к системе Коррин». Angewandte Chemie International Edition на английском языке. 3 (7): 490–496. Дои:10.1002 / anie.196404901.
- ^ Felner-Caboga, I .; Fischli, A .; Wick, A .; Pesaro, M .; Bormann, D .; Winnacker, E.L .; Эшенмозер, А. (1967). «рац-дициано- (1,2,2,7,7,12,12-гептаметилкоррин) кобальт (III)». Angewandte Chemie International Edition на английском языке. 6 (10): 864–866. Дои:10.1002 / anie.196708643.
- ^ Бенфей, О. Теодор; Моррис, Питер Дж. Т. (ред.). Роберт Бернс Вудворд - архитектор и художник в мире молекул. Серия «История современных химических наук». Филадельфия: Фонд химического наследия. ISBN 978-0941901253. ISSN 1069-2452.
- ^ а б ""Herr Woodward bedauert, daß die Sache fertig ist. "Woodward und Eschenmoser über Vitamin B12 und die Situation der organischen Chemie ". Nachrichten aus Chemie und Technik. 20 (8): 147–150. 2010. Дои:10.1002 / nadc.19720200804.
- ^ а б c d Кригер, Дж. Х. (1973). "Витамин B12: борьба за синтез ». Новости химии и машиностроения. 51 (11/12 марта): 16–29.
- ^ а б c d е ж грамм час Крейг, Дж. Уэйн (2014). "Подход Eschenmoser к витамину B12 по стратегии A / D ". Резонанс. 19 (7): 624–640. Дои:10.1007 / s12045-014-0064-4.
- ^ Смит, К. М. (1971). «Последние достижения в химии пиррольных соединений». Ежеквартальные обзоры, Химическое общество. 25: 31. Дои:10.1039 / qr9712500031.
- ^ а б c Бертеле, Эрхард; Шеффольд, Рольф; Гшвенд, Хайнц; Пезаро, Марио; Фишли, Альберт; Рот, Мартин; Шоссиг, Юрген; Эшенмозер, Альберт (2015). «Corrin Syntheses. Часть IV». Helvetica Chimica Acta. 98 (11–12): 1755–1844. Дои:10.1002 / hlca.201200342.
- ^ а б c d е ж Ямада, Ясудзи; Miljkovic, D .; Wehrli, P .; Golding, B .; Löliger, P .; Keese, R .; Müller, K .; Эшенмозер, А. (1969). «Новый тип синтеза коррина». Angewandte Chemie International Edition на английском языке. 8 (5): 343–348. Дои:10.1002 / anie.196903431. PMID 4977933.
- ^ а б c d е ж грамм час я j k Ямада, Ясудзи; Верли, Пий; Милькович, Душан; Уайлд, Ханс-Якоб; Бюлер, Никлаус; Гётчи, Эрвин; Голдинг, Бернард; Лелигер, Питер; Глисон, Джон; Пейс, Брайан; Эллис, Ларри; Хункелер, Вальтер; Шнайдер, Питер; Фюрер, Вальтер; Нордманн, Рене; Шринивасачар, Кастури; Киз, Рейнхарт; Мюллер, Клаус; Нейер, Рейнхард; Эшенмозер, Альберт (2015). «Corrin Syntheses. Часть VI». Helvetica Chimica Acta. 98 (11–12): 1921–2054. Дои:10.1002 / hlca.201500012.
- ^ а б c d Стивенс, Р. В. (1982). «Полный синтез витамина B12". В Дельфин, Д. (ред.). Витамин B12. 1. Нью-Йорк: Джон Вили и сыновья. С. 169–200. ISBN 978-0-471-03655-5.
- ^ а б c d е Уайлд, Йост (1964). Synthetische Versuche в Richtung auf natürlich vorkommende Corrinoide (PDF) (Кандидат наук). ETH Zürich (Promotionsarbeit Nr. 3492). Дои:10.3929 / ethz-a-000088927. HDL:20.500.11850/132003.
- ^ Вудворд, Р. Б. (1963). "Versuche zur Synthese des Vitamins B12". Angewandte Chemie. 75 (18): 871–872. Дои:10.1002 / ange.19630751827.
- ^ а б Вудворд, Р. Б. (1967). «Сохранение орбитальной симметрии». Ароматичность. Специальное издание химического общества. 21. Лондон: Королевское химическое общество. С. 217–249.
- ^ Деньги, Т. (1985). «Камфора: хиральный исходный материал в синтезе натуральных продуктов». Отчеты о натуральных продуктах. 2 (3): 253–89. Дои:10.1039 / np9850200253. PMID 3906448.
- ^ а б c d е ж грамм Лохер, Урс (1964). Darstellung eines Zwischenproduktes zur Synthese von Vitamin B12 (PDF) (Кандидат наук). ETH Zürich (Promotionsarbeit Nr. 3611). Дои:10.3929 / ethz-a-000090323. HDL:20.500.11850/131398.
- ^ а б c d е ж грамм час я j k Дубс, Пол (1969). Beiträge zur Synthese von Vitamin B12: Darstellung vinyloger Amidine mit der Sulfidkontraktions-Methode. (PDF) (Кандидат наук). ETH Zürich (Promotionsarbeit Nr. 4297). Дои:10.3929 / ethz-a-000093384. HDL:20.500.11850/133822.
- ^ а б c d Джексон, А. Х .; Смит, К. М. (1973). «Полный синтез пиррольных пигментов». В Апсимоне, Джон (ред.). Полный синтез натуральных продуктов. 1. С. 143–278. Дои:10.1002 / 9780470129647.ch3. ISBN 9780471032519.
- ^ а б c d е ж грамм Лелигер, Питер (1968). Darstellung eines die Ringe B und C umfassenden Zwischenproduktes zur Synthese von Vitamin B12 (PDF) (Кандидат наук). ETH Zürich (Promotionsarbeit Nr. 4074). Дои:10.3929 / ethz-a-000093406. HDL:20.500.11850/133844.
- ^ а б c Roth, M .; Dubs, P .; Götschi, E .; Эшенмозер, А. (1971). «Сульфидконтракция через алкилирующий купплунг: Eine Methode zur Darstellung von β-Dicarbonylderivaten. Über synthetische Methoden, 1. Mitteilung». Helvetica Chimica Acta. 54 (2): 710–734. Дои:10.1002 / hlca.19710540229.
- ^ а б c d е ж грамм час я j k л м п о п q р s т ты v ш Икс у z Шнайдер, Питер (1972). Полный синтез из производных дициано-кобальта (III) -5,15-бис-нор-кобиринсор-гепта-метилэфиров (PDF) (Кандидат наук). ETH Zürich (Promotionsarbeit Nr. 4819). Дои:10.3929 / ethz-a-000090603. HDL:20.500.11850/132893.
- ^ Эшенмозер, А. (1994). "B12: воспоминания и запоздалые мысли ». В Chadwick, Derek J .; Ackrill, Kate (ред.). Биосинтез тетрапиррольных пигментов. Симпозиум Фонда Ciba 180 (Симпозиум Фонда Новартис 105). Чичестер: J. Wiley & Sons. С. 309–336. ISBN 978-0471939474.
- ^ Эшенмозер, А. (1969). «Роль переходных металлов в химическом синтезе корринов». Чистая и прикладная химия. 20 (1): 1–23. Дои:10.1351 / pac196920010001.
- ^ а б c d е ж грамм час я j k л м п о п q р s т ты v ш Икс у z аа ab ac объявление ае аф аг ах Фюрер, Вальтер (1973). Totalsynthese von Vitamin B12: Der photochemische Weg (PDF) (Кандидат наук). ETH Zürich (Promotionsarbeit Nr. 5158). Дои:10.3929 / ethz-a-000086601. HDL:20.500.11850/131362.
- ^ Хубер, Дж. Ф. К. (1969). «Высокоэффективная высокоскоростная жидкостная хроматография в колонках». Журнал хроматографической науки. 7 (2): 85–90. Дои:10.1093 / chromsci / 7.2.85.
- ^ Шрайбер, Дж. (1971). "Ein Beispiel zur Anwendung der schnellen Flüssigchromatogarphie in der organischen Synthese". Chimia. 25 (12): 405–407.
- ^ Герцог, Д. (1973). "Использование жидкостной хроматографии высокого давления в органическом синтезе". Информация chimique. 119: 229–231.
- ^ а б c d е ж грамм час я j k л м п о п q р s т ты v ш Икс у z аа Мааг, Ганс (1973). Totalsynthese von Vitamin B12: Дициано-Co (III) -Cobyrinsäure-Hexamethylester-f-Amid (PDF) (Кандидат наук). ETH Zürich (Promotionsarbeit Nr. 5173). Дои:10.3929 / ethz-a-000085446. HDL:20.500.11850/131110.
- ^ а б c d е ж Вертеманн, Люциус (1968). Untersuchungen an Kobalt (II) - und Kobalt (III) -Komplexen des Cobyrinsäure-heptamethylesters (PDF) (Кандидат наук). ETH Zürich (Promotionsarbeit Nr. 4097). Дои:10.3929 / ethz-a-000093488. HDL:20.500.11850/133926.
- ^ а б c d е ж грамм час я Вудворд, Р. Б. (1979). «Синтетический витамин B12". In Zagalak, B .; Friedrich, W. (eds.). Витамин B12 (Материалы 3-го Европейского симпозиума по витамину B12 и внутренний фактор, Цюрихский университет, 5-8 марта 1979 г.). Берлин: В. де Грюйтер. С. 37–87. ISBN 3-11-007668-3.
- ^ а б Винтнер, Клод Э. (2006). "Вспоминая Институт органической химии, ETH Zürich, 1972-1990". Chimia. 60 (3): 142–148. Дои:10.2533/000942906777675029.
- ^ а б Эрнст, Людгер; Мааг, Ханс (2006). «Получение и подтверждение структуры четырех моноамидов гексаметилового эфира изомерной дицианокобириновой кислоты, несущих амидную группу на боковой цепи пропионовой кислоты». Либигс Аннален. 1996 (3): 323–326. Дои:10.1002 / jlac.199619960306.
- ^ а б c d е ж грамм Вик, Александр (1964). Untersuchungen in Richtung einer Totalsynthese von Vitamin B12 (PDF) (Кандидат наук). ETH Zürich (Promotionsarbeit Nr. 3617). Дои:10.3929 / ethz-a-000090041. HDL:20.500.11850/132537.
- ^ а б c d е ж грамм час я j k Видеркер, Рене (1968). Darstellung von Zwischenprodukten zur Synthese von Vitamin B12 (PDF) (Кандидат наук). ETH Zürich (Promotionsarbeit Nr. 4239). Дои:10.3929 / ethz-a-000087656. HDL:20.500.11850/131502.
- ^ а б c d Хубер, Вилли (1969). Beiträge zur Synthese von Vitamin B12: Zum Problem der (C-D) -Verknüpfung (PDF) (Кандидат наук). ETH Zürich (Promotionsarbeit Nr. 4298). Дои:10.3929 / ethz-a-000090323. HDL:20.500.11850/132700.
- ^ а б c d е ж грамм час я j k л м п Шиллинг, Вальтер (1974). Totalsynthese von Vitamin B12. Darstellung von Zwischenprodukten und partialsynthetische Endstufen (PDF) (Кандидат наук). ETH Zürich (Promotionsarbeit Nr. 5352). Дои:10.3929 / ethz-a-000085344. HDL:20.500.11850/131064.
- ^ а б c Эшенмозер, Альберт (2015). «Вступительные замечания по серии публикаций Corrin Syntheses-части I-VI'". Helvetica Chimica Acta. 98 (11–12): 1475–1482. Дои:10.1002 / hlca.201400399.
- ^ Шеффольд, Рольф; Бертеле, Эрхард; Гшвенд, Хайнц; Хойзерманн, Вернер; Верли, Пий; Хубер, Вилли; Эшенмозер, Альберт (2015). «Corrin Syntheses. Часть II». Helvetica Chimica Acta. 98 (11–12): 1601–1682. Дои:10.1002 / hlca.201200095.
- ^ Пезаро, Марио; Эльсингер, Фриц; Боос, Гельмут; Фельнер-Кабога, Иво; Гриби, Ханспетер; Вик, Александр; Гшвенд, Хайнц; Эшенмозер, Альберт (2015). «Corrin Syntheses. Часть III». Helvetica Chimica Acta. 98 (11–12): 1683–1754. Дои:10.1002 / hlca.201200308.Блазер, Ганс-Ульрих; Виннакер, Эрнст-Людвиг; Фишли, Альберт; Хардеггер, Бруно; Борман, Дитер; Хашимото, Наото; Шоссиг, Юрген; Киз, Рейнхарт; Эшенмозер, Альберт (2015). "Corrin Syntheses. Часть V". Helvetica Chimica Acta. 98 (11–12): 1845–1920. Дои:10.1002 / hlca.201300064.
- ^ «Исследовательская коллекция ETH (ранее электронная коллекция ETH)». ETH Цюрих. Получено 2020-01-25.
- ^ Гото, Тошио (1975). "Глава 11.34: Синтез витамина B12". В Наканиши, Кодзи; Гото, Тошио; Шо, Ито; Натори, Шинсаку; Нозоэ, Шигео (ред.). Химия натуральных продуктов. 2. Токио: Коданша / Academic Press. С. 480–496. ISBN 0-12-513902-0.
- ^ Ритер, Дорис; Мульцер, Иоганн (2003). «Полный синтез кобирной кислоты: историческое развитие и последние синтетические инновации». Европейский журнал органической химии. 2003: 30–45. Дои:10.1002 / 1099-0690 (200301) 2003: 1 <30 :: AID-EJOC30> 3.0.CO; 2-I.
- ^ Кори, Э. Дж.; Чоу, С. У .; Шеррер, Р. А. (1957). «Синтез α-санталена и транс-Δ11,12-изо-α-сантален ". Журнал Американского химического общества. 79 (21): 5773–5777. Дои:10.1021 / ja01578a049.Guha, P.C .; Бхаттачаргья, С. К. (1944). «Санталол серии. II. Синтез d- и дл-π-гидроксикамфора, d- и дл-тересанталол и d- и дл-трициклоэкзанталовая кислота ». Журнал Индийского химического общества. 21: 271–280.Кори, Э. Дж.; Оно, Масаджи; Чоу, С. У .; Шеррер, Роберт А. (1959). «Катализируемое кислотой расщепление π-замещенных трицикленов. Синтез 3,8-циклокамфора». Журнал Американского химического общества. 81 (23): 6305–6309. Дои:10.1021 / ja01532a048.Хассельстрем, Торстен (1931). «Исследования производных π-камфары. II. Идентичность дигидро-терезанталовой кислоты с 7-π-апокамфанкарбоновой кислотой». Журнал Американского химического общества. 53 (3): 1097–1103. Дои:10.1021 / ja01354a043.
- ^ Каски, Б.А. (1971). Исследования по упаковке в молекулярных кристаллах (Кандидат наук). Гарвардский университет. С. II-1.
- ^ Блазер, Ганс-Ульрих (1971). Herstellung und Eigenschaften eines Metallfreien Corrin-Derivates (PDF) (Кандидат наук). ETH Zürich (Promotionsarbeit Nr. 4662). Дои:10.3929 / ethz-a-000091385. HDL:20.500.11850/133210.
- ^ Фишли, Альберт (1968). Die Synthese Metallfreier Corrine (PDF) (Кандидат наук). ETH Zürich (Promotionsarbeit Nr. 4077). Дои:10.3929 / ethz-a-000267791. HDL:20.500.11850/137445.
- ^ Jauernig, D .; Rapp, P .; Руофф, Г. (1973). «5-нор-, 15-нор- и 5,15-диноркориноид». Hoppe-Seyler's Zeitschrift für Physiologische Chemie. 354 (8): 957–966.
- ^ Manasse, O .; Самуэль, Э. (1902). "Reactionen des Campherchinons". Berichte der Deutschen Chemischen Gesellschaft. 35 (3): 3829–3843. Дои:10.1002 / cber.190203503216.
- ^ Wick, A.E .; Феликс, Дороти; Стин, Катарина; Эшенмозер, А. (1964). "Claisen'sche Umlagerungen bei Allyl- und Benzylalkoholen mit Hilfe von Acetalen des N, N-Диметилацетамиды. Vorläufige Mitteilung ". Helvetica Chimica Acta. 47 (8): 2425–2429. Дои:10.1002 / hlca.19640470835.Феликс, Дороти; Гшвенд-Стин, Катарина; Wick, A.E .; Эшенмозер, А. (1969). "Claisen'sche Umlagerungen bei Allyl- und Benzylalkoholen mit 1-Dimethylamino-1-metxy-äthen". Helvetica Chimica Acta. 52 (4): 1030–1042. Дои:10.1002 / hlca.19690520418.
- ^ Джонсон, Уильям Саммер; Вертеманн, Люциус; Бартлетт, Уильям Р .; Brocksom, Тимоти Дж .; Ли, Цунг-Ти; Фолкнер, Д. Джон; Петерсен, Майкл Р. (1970). «Простая стереоселективная версия перегруппировки Клейзена, приводящая к транс-тризамещенным олефиновым связям. Синтез сквалена». Журнал Американского химического общества. 92 (3): 741–743. Дои:10.1021 / ja00706a074.Ирландия, Роберт Э .; Мюллер, Ричард Х .; Уиллард, Элвин К. (1976). «Перегруппировка енолята Клайзена сложного эфира. Стереохимический контроль посредством стереоселективного образования енолята». Журнал Американского химического общества. 98 (10): 2868–2877. Дои:10.1021 / ja00426a033.
- ^ Уайлд, Ханс-Якоб (1972). Die Synthese von Corrin-Komplexen durch photochemische A / D-Cycloisomerisierung (PDF) (Кандидат наук). ETH Zürich (Promotionsarbeit Nr. 4848). Дои:10.3929 / ethz-a-000090212. HDL:20.500.11850/132648.
- ^ Гардинер, Морин; Томсон, Эндрю Дж. (1974). «Люминесцентные свойства некоторых синтетических металлокорринов». Журнал химического общества, Dalton Transactions (8): 820. Дои:10.1039 / DT9740000820.
- ^ Виннакер, Эрнст-Людвиг (1968). Ligandreaktivität synthetischer Kobalt (III) -Corrin-Komplexe (PDF) (Кандидат наук). ETH Zürich (Promotionsarbeit Nr. 4177). Дои:10.3929 / ethz-a-000150375. HDL:20.500.11850/136417. Cite имеет пустой неизвестный параметр:
|1=(помощь) - ^ Bonnett, R .; Годфри, Дж. М .; Математика, В. Б. (1971). «Циано-13-эпикобаламин (Неовитамин В12) и его родственники ». Журнал химического общества C: Органический. 22: 3736–43. Дои:10.1039 / j39710003736. PMID 5167083.
- ^ Kempe, U. M .; Дас Гупта, Т.К .; Blatt, K .; Gygax, P .; Феликс, Дороти; Эшенмозер, А. (1972). "α-Хлор-нитрон I: Darstellung und Ag+-induzierte Reaktion mit Olefinen. Über synthetische Methoden, 5. (Vorläufige) Mitteilung ". Helvetica Chimica Acta. 55 (6): 2187–2198. Дои:10.1002 / hlca.19720550640.