Мышечная дистрофия конечностей и пояса - Limb–girdle muscular dystrophy
| Мышечная дистрофия конечностей и пояса | |
|---|---|
| Другие имена | Мышечная дистрофия Эрба[1] |
 | |
| Белок MYOT (также известный как TTID, один из многих генов, мутации которых ответственны за это состояние) | |
| Специальность | Физическая медицина и реабилитация |
| Симптомы | Псевдогипертрофия[2] |
| Типы | LGMD1 и LGMD2[3] |
| Диагностический метод | Иммуногистохимический дистрофин тесты[4] |
| Уход | Профессиональная, логопедическая и физиотерапия[5] |
Мышечная дистрофия конечностей и пояса или (LGMD) представляет собой генетически и клинически гетерогенную группу редких мышечные дистрофии.[6] Для него характерны прогрессивные атрофия мышц поражает преимущественно мышцы бедер и плеч. LGMD имеет аутосомный образец наследство и в настоящее время не известно лекарства или лечения.[7][8]
Признаки и симптомы
Симптомами человека с мышечной дистрофией конечностей и поясов (LGMD) обычно являются большие трудности при ходьбе, подъеме и спуске по лестнице и вставании со стула. Также присутствует неспособность наклониться или присесть. Из-за этих трудностей падение может происходить часто. Подъем определенных предметов, а также трудности с вытягиванием рук за голову или над головой варьируются от трудного до невозможного в зависимости от степени тяжести. В конце концов, способность ходить и бегать ухудшается.[2][4]
В дальнейшем презентации человек с LGMD может иметь:

- Псевдогипертрофия[2]
- Мышцы гипертрофия[9]
- Проблемы с дыхательными мышцами[9]
- Низкий назад дискомфорт[2]
- Сердцебиение[2]
- Проблемы с дистальными мышцами[9]
- Лицевая мышца слабое место[2]
- Слабый плечо мышца [2]
Заболевание со временем неизбежно ухудшается, хотя у одних пациентов прогрессирование происходит быстрее, чем у других. Со временем болезнь может поразить и другие мышцы, например, расположенные на лице. Заболевание обычно приводит к зависимости от инвалидной коляски в течение нескольких лет после появления симптомов, но существует высокая вариабельность между пациентами, при этом некоторые пациенты сохраняют подвижность.[8][2]
Слабость мышц обычно симметрична, проксимальный, и медленно прогрессирующий. В большинстве случаев при LGMD боль отсутствует, и психическая функция не нарушается. LGMD может начаться в детстве, подростковом возрасте, молодом взрослом возрасте или даже позже, возраст начала обычно составляет от 10 до 30. Оба пола страдают одинаково, когда мышечная дистрофия конечностей начинается в детстве, прогрессирование кажется более быстрым, а болезнь более тяжелой. отключение. Когда расстройство начинается в подростковом или взрослом возрасте, болезнь, как правило, не столь тяжелая и прогрессирует медленнее. При поступлении нет сенсорной нейропатии, вегетативной или висцеральной дисфункции.[требуется медицинская цитата ]
Генетика
С точки зрения генетики LGMD - это унаследованный расстройство, хотя оно может передаваться по наследству доминирующий или рецессивный генетический дефект. Результатом дефекта является то, что мышцы не могут правильно формировать определенные белки необходим для нормальной работы мышц. Могут быть затронуты несколько различных белков, и конкретный белок, который отсутствует или является дефектным, определяет конкретный тип мышечной дистрофии. Среди белков, на которые влияет LGMD, входят α, β, γ и δ саркогликаны. В саркогликанопатии возможно, поддается генная терапия.[3]
Диагностика
Диагноз мышечной дистрофии конечностей и пояса может быть поставлен с помощью мышечной ткани. биопсия, который покажет наличие мышечной дистрофии, а генетическое тестирование используется, чтобы определить, какой тип мышечной дистрофии у пациента. Иммуногистохимический тесты на дистрофин могут указывать на снижение уровня дистрофина, обнаруженное в саркогликанопатии. С точки зрения саркогликан дефицит может быть дисперсией (если α-саркогликан и γ-саркогликан отсутствуют, значит, в LGMD2D есть мутация).[4]
2014 год Резюме рекомендаций, основанных на фактах: Диагностика и лечение дистрофий конечностей и поясов и дистрофий указывает на то, что лицам с подозрением на наследственное заболевание следует пройти генетическое тестирование. Другие тесты / анализы:[4][5]
- Высокий уровень КФК (в 10-150 раз больше нормы)
- МРТ может указывать на разные типы LGMD.
- ЭМГ может подтвердить миопатическую характеристику заболевания.
- Электрокардиография (могут возникнуть сердечные аритмии при LGMD1B)
Типы
Семейство «LGMD1» - это аутосомно-доминантный, а семейство "LGMD2" аутосомно-рецессивный.[8] Мышечная дистрофия конечностей и поясов объясняется с точки зрения гена, локуса, OMIM и типа следующим образом:
| Семья | Наследование | Тип | OMIM | Ген | Locus | Примечания |
|---|---|---|---|---|---|---|
| LGMD1 | аутосомно-доминантный  138 пикселей | LGMD1A | 159000 | TTID | ||
| LGMD1B | 159001 | LMNA | ||||
| LGMD1C | 607801 | CAV3 | ||||
| LGMD1D | 603511 | ДНКJB6 | ||||
| LGMD1E | 601419 | DES | ||||
| LGMD1F | 608423 | TNPO3 | 7q32.1 – q32.2 | |||
| LGMD1G | 609115 | HNRPDL | 4q21 | |||
| LGMD1H | 613530 | 3п25.1 – п23 | ||||
| LGMD2 | аутосомно-рецессивный  | LGMD2A | 253600 | CAPN3 | Часто называют «кальпаинопатией».[10] | |
| LGMD2B | 253601 | DYSF | LGMD с мутацией в этом гене, наряду с миопатией Миёси типа 1 (MMD1 - 254130 ) иногда называют дисферлинопатиями.[11] | |||
| LGMD2C | 253700 | SGCG | ||||
| LGMD2D | 608099 | SGCA | ||||
| LGMD2E | 604286 | SGCB | ||||
| LGMD2F | 601287 | SGCD | ||||
| LGMD2G | 601954 | TCAP | ||||
| LGMD2H | 254110 | TRIM32 | ||||
| LGMD2I | 607155 | ФКРП | ||||
| LGMD2J | 608807 | TTN | ||||
| LGMD2K | 609308 | POMT1 | ||||
| LGMD2L | 611307 | АНО5 | ||||
| LGMD2M | 611588 | FKTN | ||||
| LGMD2N | 607439 | POMT2 | ||||
| LGMD2O | 606822 | POMGNT1 | ||||
| LGMD2Q | 613723 | PLEC1 |
Уход

Есть несколько исследований, подтверждающих эффективность упражнение при мышечной дистрофии конечностей и поясов. Однако исследования показали, что упражнения могут необратимо повредить мышцы из-за интенсивного сокращения мышц.[12] Физиотерапия может потребоваться для поддержания максимальной силы мышц и гибкости суставов. Суппорта может использоваться для поддержания мобильности и качества жизни. Требуется пристальное внимание к здоровью легких и сердца, кортикостероиды у пациентов с LGMD 2C-F показывают некоторое улучшение.[9] Дополнительно люди могут следить управление что следует:[5]
- Профессиональные терапия
- Респираторный терапия
- Логопедия
- Нейтрализующие антитела к миостатин не следует преследовать
С точки зрения прогноза мышечной дистрофии конечностей и поясов в ее самой легкой форме, пораженные люди обладают почти нормальной мышечной силой и функцией. LGMD обычно не является смертельным заболеванием, хотя в конечном итоге может ослабить сердце и респираторный мышцы, приводящие к болезни или смерти из-за вторичных нарушений. Частота мышечной дистрофии конечностей и поясов колеблется от 1 на 14 500 (в некоторых случаях 1 на 123 000).[6][8]
Исследование
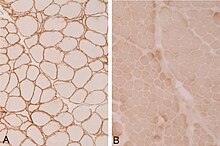
В настоящее время проводится множество исследований, направленных на различные формы мышечной дистрофии конечностей и поясов. Среди методов, которые считаются многообещающими для лечения, можно назвать терапию с переносом генов,[13] который работает, вставляя в клетки дефектные гены со здоровым геном.[14]
Согласно обзору Bengtsson et al. некоторый успех генной терапии, опосредованной AAV (при различных заболеваниях), повысил интерес исследователей, CRISPR / Cas9 и пропуск экзонов способствует достижению этих терапевтических целей. Мышечные дистрофии конечностей и поясов имеют много разных типов, которые возникают из-за разных генных мутаций. LGMD2D вызывается мутацией в гене α-саркогликана. В будущем лечение может быть обеспечено генной терапией с помощью рекомбинантных адено -ассоциированные векторы.[15]
Напротив, согласно обзору Straub, et al. есть несколько исследовательских проблем, на которые необходимо обратить внимание, редкость заболевания, наше плохое понимание механизма LGMD2 и отсутствие когорт пациентов, следовательно, биомаркеры для лиц с LGMD2 отсутствуют. Далее в обзоре утверждается, что модели LGMD2 на животных использовались для анализа терапевтических препаратов. Также добавив, что пока преднизон использовался и оказал положительное влияние на пациентов с LGMD2, до сих пор нет доказательств его эффективности в плацебо-контролируемых испытаниях.[16]
Смотрите также
Рекомендации
- ^ Ньюфаундленд, FRCP Уильям Прайс-Филлипс, доктор медицины, FRCP (C) Медицинский факультет Центр медицинских наук Мемориальный университет Ньюфаундленда Сент-Джонс (2009-05-06). Компаньон клинической неврологии. Oxford University Press, США. п. 579. ISBN 9780199710041.
- ^ а б c d е ж грамм час Энциклопедия MedlinePlus: Мышечные дистрофии конечностей и пояса
- ^ а б Справка, Дом генетики. «пояснично-конечностная мышечная дистрофия». Домашний справочник по генетике. Получено 2016-04-22.
- ^ а б c d «Мышечная дистрофия конечностей и поясов: основы практики, предыстория, патофизиология». Август 2018. Цитировать журнал требует
| журнал =(Помогите) - ^ а б c Нараянасвами, Пушпа; Вайс, Майкл; Селцен, Дуйгу; Дэвид, Уильям; Рейнор, Элизабет; Картер, Грегори; Виклунд, Мэтью; Барон, Ричард Дж .; Энсруд, Эрик (2014-10-14). «Резюме руководства, основанное на фактах: Диагностика и лечение дистрофии конечностей и дистальных отделов». Неврология. 83 (16): 1453–1463. Дои:10.1212 / WNL.0000000000000892. ISSN 0028-3878. ЧВК 4206155. PMID 25313375.
- ^ а б «Конечностно-поясная мышечная дистрофия».
- ^ «Лечение и лечение конечностно-поясной мышечной дистрофии: медицинское обслуживание, хирургическое лечение, консультации». Август 2018. Цитировать журнал требует
| журнал =(Помогите) - ^ а б c d Пегораро, Елена; Хоффман, Эрик П. (1993-01-01). Пагон, Роберта А .; Адам, Маргарет П .; Ardinger, Holly H .; Уоллес, Стефани Э .; Амемия, Энн; Бин, Лора JH; Bird, Thomas D .; Фонг, Чин-То; Меффорд, Хизер С. (ред.). Обзор мышечной дистрофии конечностей и пояса. Сиэтл (Вашингтон): Вашингтонский университет, Сиэтл. PMID 20301582.обновление 2012
- ^ а б c d "Конечностно-поясная мышечная дистрофия | Доктор".
- ^ Lasa-Elgarresta, J; Москейра-Мартин, L; Налдаиз-Гастеси, N; Саенс, А; Лопес де Мунайн, А; Вальехо-Илларраменди, А (13 сентября 2019 г.). «Кальциевые механизмы при мышечной дистрофии конечностей и пояса с CAPN3 Мутации ». Международный журнал молекулярных наук. 20 (18): 4548. Дои:10.3390 / ijms20184548. ЧВК 6770289. PMID 31540302.
- ^ Аоки, Масаси (5 марта 2015 г.). «Дисферлинопатия». GeneReviews.
- ^ Сицилиано Дж., Симончини С., Джаннотти С., Зампа В., Анджелини С., Риччи Дж. (2015). «Мышечные упражнения при мышечных дистрофиях пояса конечностей: подводные камни и преимущества». Acta Myologica. 34 (1): 3–8. ЧВК 4478773. PMID 26155063.
- ^ Представитель по связям с общественностью, Мелани Мартинес, Управление по связям с общественностью. «Конечностно-поясная мышечная дистрофия». www.niams.nih.gov. Получено 2016-04-22.
- ^ Справка, Дом генетики. "Как работает генная терапия?". Домашний справочник по генетике. Получено 2016-04-23.
- ^ Bengtsson, Niclas E .; Сето, Джейн Т .; Холл, Джон К .; Чемберлен, Джеффри С .; Одом, Гай Л. (15.04.2016). «Ход и перспективы клинических исследований генной терапии мышечных дистрофий». Молекулярная генетика человека. 25 (R1): R9 – R17. Дои:10.1093 / hmg / ddv420. ISSN 0964-6906. ЧВК 4802376. PMID 26450518.
- ^ Штрауб, Фолькер; Бертоли, Марта (01.02.2016). «Где мы находимся в испытании готовности к аутосомно-рецессивным мышечным дистрофиям пояса конечностей?». Нервно-мышечные расстройства. 26 (2): 111–125. Дои:10.1016 / j.nmd.2015.11.012. PMID 26810373. S2CID 23787096. - через ScienceDirect (Может потребоваться подписка или контент может быть доступен в библиотеках.)
дальнейшее чтение
- Котта, Ана; Карвалью, Эльмано; да-Кунья-Жуниор, Антонио Лопес; Пайм, Джулия Филарди; Navarro, Monica M .; Валичек, Жаклин; Менезеш, Мириам Мело; Нуньес, Симоне Вилела; Ксавье Нето, Рафаэль (2014). «Дифференциальная диагностика распространенных рецессивных мышечных дистрофий пояса конечностей: почему и как?». Arquivos de Neuro-Psiquiatria. 72 (9): 721–734. Дои:10.1590 / 0004-282X20140110. ISSN 0004-282X. PMID 25252238.
- Лю, Цзянь; Харпер, Скотт К. (2012-08-01). «Генная терапия на основе РНКи для мышечных дистрофий доминирующего пояса конечностей». Современная генная терапия. 12 (4): 307–314. Дои:10.2174/156652312802083585. ISSN 1566-5232. ЧВК 4120526. PMID 22856606.
- АНЖЕЛИНИ, КОРРАДО; ТАСКА, ЭЛИСАБЕТТА; НАСЧИМБЕНИ, АННА ЧИАРА; ФАНИН, МАРИНА (01.12.2014). «Мышечная усталость, nNOS и атрофия мышечных волокон при мышечной дистрофии пояса конечностей». Acta Myologica. 33 (3): 119–126. ISSN 1128-2460. ЧВК 4369848. PMID 25873780.
внешняя ссылка
| Классификация | |
|---|---|
| Внешние ресурсы |
| Scholia имеет тема профиль для Конечностно-поясная мышечная дистрофия. |
| Типы | |
|---|---|
| Национальные / международные организации |
|
| Национальные / международные мероприятия |
|
| Клинические испытания | |