Синдром Ваарденбурга - Waardenburg syndrome
| Синдром Ваарденбурга | |
|---|---|
| Другие имена | Синдром Клейна – Ваарденбурга (тип 3), синдром Шаха – Ваарденбурга (тип 4) |
 | |
| Черты лица синдрома Ваарденбурга 1 типа (от Ян ван дер Хёв описание, 1916 г.) | |
| Специальность | Медицинская генетика |
Синдром Ваарденбурга группа редких генетические условия характеризуется по крайней мере некоторой степенью врожденной потери слуха и дефицита пигментации, которые могут включать ярко-голубые глаза (или один голубой глаз и один карие глаз ), а белый чуб или участки светлой кожи. Эти основные характеристики составляют 2-й тип состояния; в типе 1 также имеется более широкий промежуток между внутренними уголками глаз, который называется телекантус, или dystopia canthorum.[1] При типе 3, который встречается редко, руки и кисти также деформированы, с постоянные контрактуры пальцев или сросшиеся пальцы, в то время как в типе 4 у человека также есть Болезнь Гиршпрунга, что является врожденным недостатком нервов в кишечнике, приводящим к дисфункция кишечника.[2][3] Также существует как минимум два типа (2E и PCWH), которые могут привести к Центральная нервная система такие симптомы, как задержка развития и мышечный тонус аномалии.[4]
Синдром вызван мутациями в любом из нескольких генов, влияющих на деление и миграция из клетки нервного гребня в течение эмбриональное развитие (хотя некоторые из задействованных генов также влияют на нервная трубка ).[5] Клетки нервного гребня стволовые клетки осталось после закрытие нервной трубки, которые продолжают формировать различные клетки не центральной нервной системы в разных частях тела, в том числе меланоциты, различные кости и хрящи лица и внутреннее ухо и периферийные нервы кишечника.[6] Тип 1 вызван мутацией в PAX3 ген, в то время как ген, который чаще всего вызывает тип 2 при мутации, MITF.[1][7] Тип 3 является более серьезным проявлением типа 1 и вызван мутацией в том же гене, а тип 4 чаще всего вызывается мутацией в том же гене. SOX10.[2][8] Мутации в других генах также могут вызывать различные типы, и некоторым из них были присвоены собственные подтипы, обозначенные буквами. Большинство типов аутосомно-доминантный.
По оценкам, распространенность синдрома Ваарденбурга составляет 1 случай на 42000.[5][8] Типы 1 и 2 являются наиболее распространенными, составляя примерно половину и треть случаев соответственно, в то время как тип 4 составляет пятый, а тип 3 - менее 2% случаев.[8] По оценкам, 2–5% врожденно глухих людей страдают синдромом Ваарденбурга.[8] Описание синдрома относится как минимум к первой половине 20 века, однако он назван в честь голландского офтальмолога и генетика. Петрус Йоханнес Ваарденбург, который описал это в 1951 году.[9][10] Его подтипы постепенно открывались в последующие десятилетия, и в основном им приписывали гены в 1990-х и 2000-х годах.
Признаки и симптомы
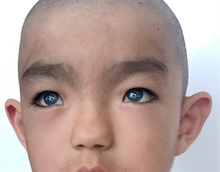
Синдром Ваарденбурга имеет несколько разных типов с некоторыми вариациями симптомов, и симптомы могут различаться у людей того же типа. Две особенности, характерные для всех типов синдрома Ваарденбурга, в некоторой степени являются врожденными. нейросенсорная тугоухость и некоторая степень дефицита пигментации, чаще всего в глазах.[11]
Тип 1
Тип 1 характеризуется врожденным нейросенсорная тугоухость, пигментные недостатки волос, такие как белая прядь волос (полиоз ) в передней центральной части головы или преждевременное поседение, пигментный дефицит глаз, такой как глаза разного цвета (полный гетерохромия иридум ), множественные цвета в глазу (секторная гетерохромия радужной оболочки) или блестящие голубые глаза, участки депигментации кожи и более широкий промежуток между внутренними уголками глаз, называемый телекантус, или dystopia canthorum. Другие черты лица, связанные с типом 1, могут включать высокую переносицу, плоский кончик носа, монобровь (синофрисы), меньшие края ноздрей (крылья) или гладкие желобок.[1]
Тип 2
Различие, которое отличает тип 2 от типа 1, заключается в том, что у пациентов нет более широкого промежутка между внутренними уголками глаз (telecanthus / dystopia canthorum). Нейросенсорная тугоухость, как правило, встречается чаще и тяжелее при этом типе.[7][12] Безусловно, наиболее распространенным геном, вызывающим этот тип при мутации, является MITF (классифицируется как тип 2А). Если у двух человек с мутацией в этом гене есть ребенок, несущий обе мутации (гомозиготный ), вероятность которой составляет 25%, у ребенка присутствуют дополнительные симптомы, такие как отверстие в радужной оболочке (колобома ), маленькие глаза (микрофтальм ), окаменелые кости (остеопетроз ), макроцефалия, альбинизм и глухота.[7]
Есть два известных пациента с мутациями в обеих копиях SNAI2 (классифицируется как тип 2D); у этих людей был синдром Ваарденбурга типа 2, но не было нарушений пигментации волос.[13]
Когда синдром Ваарденбурга типа 2 вызван мутацией в SOX10 (классифицируется как тип 2E), в некоторых случаях он может проявляться множественными неврологическими симптомами. Они могут включать задержку развития, раннее детство. нистагм, повышенный мышечный тонус, белое вещество аномалии или гипомиелинизация в мозгу, аутичный -подобное поведение и недоразвитость или полное отсутствие многих внутреннее ухо структуры, такие как вестибулярный аппарат или улитка. Отсутствие обоняния (аносмия ) из-за отсутствия обонятельная луковица в головном мозге также может присутствовать.[4]
Тип 3
Также известен как Синдром Клейна – Ваарденбурга, или синдром Ваарденбурга-Клейна, тип 3 имеет те же симптомы, что и тип 1 (и вызывается мутациями в том же гене), но имеет дополнительные симптомы, которые влияют на руки и кисти. Они могут включать совместные контрактуры пальцев (камптодактилия ), из-за недоразвитых мышц, а также сросшихся пальцев (синдактилия ) или крылатые лопатки. Возможны также микроцефалия и задержка развития.[2]
Тип 4
Также известен как Синдром Шаха-Ваарденбурга, или синдром Ваарденбурга-Шаха, тип 4 имеет большинство тех же характеристик, что и тип 2 (то есть нет телекантуса или очевидного более широкого глазного щита), но с добавлением Болезнь Гиршпрунга, что является врожденным недостатком нервов в кишечнике, приводящим к дисфункции кишечника.[3][14] Кроме того, потеря слуха встречается не так часто, как при типе 2.[3] Редко, заячья губа Сообщалось о такой форме синдрома Ваарденбурга.[15]
Тип 4 также может быть вызван мутацией в SOX10 (тот же ген, что и у типа 2E), в котором он известен как тип 4C; Потеря слуха у этого типа очень распространена и очень серьезна.[16]
PCWH
Мутация в SOX10, ген, участвующий в типах 2E и 4C, иногда может вызывать симптомы обоих типов (неврологические симптомы, как иногда наблюдается при типе 2E, и болезнь Гиршпрунга, как показано в типе 4). Когда это происходит, это называется периферическая демиелинизирующая нейропатия - центральная дисмиелинизирующая лейкодистрофия - синдром Ваарденбурга - болезнь Гиршпрунга (PCWH).[17][18]
Причина

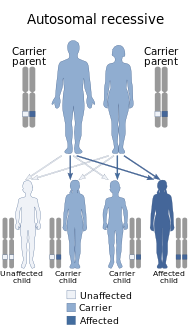
Синдром Ваарденбурга вызывается мутациями в любом из нескольких генов, влияющих на работу нервный гребень клетки в эмбриональное развитие. Большинство типов синдрома Ваарденбурга вызваны: аутосомно-доминантный мутации. Аутосомно-рецессивные редки. В большинстве случаев заболевший унаследовал его от одного из родителей с одной из доминирующих форм заболевания. Небольшой процент случаев является результатом спонтанных новых мутаций в гене, когда семейная история этого заболевания отсутствует.
Нервный гребешок - это группа временных мигрирующих клеток, которые остаются после нервная трубка закрылся (нейруляция ), примерно на четвертой неделе эмбрионального развития. Они несут ответственность за дифференциацию в различные группы клеток, которые достигают разных областей тела. Нервная трубка и нервный гребень происходят от эктодерма; нервная трубка продолжает формировать мозг и спинной мозг, в то время как клетки нервного гребня в конечном итоге продолжают формировать различные кости и хрящи черепа и лица, мигрируя через глоточные дуги. Они также дифференцируются на сосудистая полоска из улитка, нервы и глия кишечника (мышечно-кишечное сплетение ), Шванновские клетки, который миелиновый то периферическая нервная система чтобы обеспечить достаточную проводимость, одонтобласты, которые производят дентин глубоко в зубах, некоторые нейроэндокринные клетки, соединительная ткань вокруг слюна, слезный, гипофиз, вилочковая железа и щитовидная железа железы, соединительная ткань глаза, например строма радужной оболочки и роговица и трабекулярная сеть,[19] и меланоциты, в том числе те, которые находятся в строме радужной оболочки, которые вызывают коричневый цвет глаз через меланин. Клетки нервного гребня также играют роль в формировании мышц, включая мышцу стенки определенных сердечных артерий.[6]
Причины подтипов
- Тип 1 вызван аутосомно-доминантной мутацией в гене PAX3.[1] PAX3, или парная коробка 3, это фактор транскрипции который играет роль в поддержании открытого окна времени для определенных клеток нервного гребня (например, клеток головы и глаз) для деления и миграции до того, как они терминальная дифференциация (т.е. поддерживать их в стволовая клетка штат). Таким образом, мутации в этом гене преждевременно останавливают их деление и миграцию, что приводит к незначительному недостатку развития определенных хрящей и костей лица, а также к недоразвитию структур внутреннего уха и недостатку меланоцитов в строме радужки. Имеются данные о том, что PAX3 также регулирует клетки до образования нервного гребня, то есть нервной трубки, поскольку мыши с мутациями потери функции в одной из копий PAX3 имеют дефекты нервной трубки такие как расщелина позвоночника или экзэнцефалия.[5]
- Тип 2 вызывается мутацией в любом из ряда генов, наиболее распространенным из которых является MITF, когда он относится к типу 2А.
- Тип 2А вызван аутосомно-доминантной мутацией в гене MITF.[7] MITF, или фактор транскрипции, связанный с микрофтальмией, представляет собой фактор транскрипции, который играет более специализированную роль в нервном гребне и более строго участвует после формирования нервного гребня (было обнаружено, что PAX3 и SOX10 активируют MITF).[20] Известно, что меланоциты остеокласты, тучные клетки и пигментный эпителиальный сетчатка клетки делиться и мигрировать. Участие в остеокластах объясняет, почему мутации в обеих копиях MITF может привести к затвердеванию костей (остеопетроз ), поскольку остеокласты отвечают за разрушение костей. MITF также активирует транскрипцию тирозиназа, фермент, который выполняет первый шаг в создании меланина (окисляющий тирозин ). Мутация в копии MITF также может привести к Синдром Тиц, который отличается от синдрома Ваарденбурга равномерным альбинизмом вместо пятнистой депигментации.[5]
- Тип 2B вызывается аутосомно-доминантной мутацией неизвестного гена на хромосоме 1 в диапазоне локусов 1p21–1p13.3. Ген был условно назван WS2B.[21][22]
- Тип 2C вызывается аутосомно-доминантной мутацией неизвестного гена на хромосоме 8 в локусе 8p23. Ген был условно назван WS2C.[23][24]
- Тип 2D вызван аутосомно-рецессивной мутацией в обеих копиях гена. SNAI2. В исследовании, обнаружившем эту связь, было обнаружено, что SNAI2 активируется MITF как часть развития нервного гребня, и это объясняет, почему мутации в MITF вызывают синдром Ваарденбурга, так как это приводит к отсутствию активации SNAI2. Мутации в единственном экземпляре SNAI2 также было обнаружено, что они вызывают пятна депигментации волос (пегий ) без каких-либо других симптомов.[25]
- Тип 2E вызван аутосомно-доминантной мутацией в гене SOX10.[4]
- В редких случаях мутация в гене, отличном от тех, которые известны в настоящее время, может быть причиной синдрома Ваарденбурга с признаками типа 2. Обычно он классифицируется как просто тип 2, но может получить свой собственный подтип после того, как ген или локус идентифицирован и установлен. .[7]
- Тип 3 вызван мутацией в гене PAX3, тот же ген, что и у типа 1.[2] Он может передаваться по аутосомно-доминантному или аутосомно-рецессивному типу; у двух родителей с синдромом Ваарденбурга типа 1 может быть ребенок, несущий обе мутировавшие копии PAX3 ген (вероятность 25%) и присутствует с синдромом Ваарденбурга типа 3. A миссенс-мутация было зарегистрировано, чтобы иметь такой эффект. Тем не менее, синдром Ваарденбурга типа 3 также может проявляться спонтанно только с одной мутированной копией PAX3. Удаление парный домен задокументировано, что область гена оказывает такой эффект.[26][5] Однако серьезной корреляции между типом мутации и тяжестью заболевания не обнаружено. Серьезность обычно определяется мутациями в других генах (эпистаз ), о чем свидетельствуют различные семейные паттерны тяжести, не связанные с типом мутации Ваарденбурга.[5] Мутации в обеих копиях PAX3 иногда приводили к смерти до или вскоре после рождения, и мыши с мутациями потери функции в обеих копиях гена не выживают.[5]
- Тип 4 вызывается мутацией в любом из ряда генов, наиболее распространенным из которых является SOX10, когда он классифицируется как тип 4С.
Исследование было проведено на редком случае двойного гетерозиготного ребенка, у каждого родителя которого были только одиночные мутации в MITF или PAX3. Эффект двойных гетерозиготных мутаций в генах MITF и PAX3 в WS1 и WS2 может усиливать симптомы поражения пигмента. Это приводит к выводу, что двойная мутация MITF связана с конечностью синдрома Ваарденбурга и может влиять на фенотипы или симптомы синдрома.[28]
Таблица классификации
| Тип | OMIM | Ген | Locus | Наследование |
|---|---|---|---|---|
| Тип 1 (WS1) | 193500 | PAX3 | 2q36.1[29] | Аутосомно-доминантный |
| Тип 2A (WS2A, первоначально WS2) | 193510 | MITF | 3п14.1 – п12.3 | Аутосомно-доминантный |
| Тип 2B (WS2B) | 600193 | WS2B | 1p21 – p13.3 | Аутосомно-доминантный |
| Тип 2C (WS2C) | 606662 | WS2C | 8p23 | Аутосомно-доминантный |
| Тип 2D (WS2D) | 608890 | SNAI2 | 8q11 | Аутосомно-рецессивный |
| Тип 2E (WS2E) | 611584 | SOX10 | 22q13.1 | Аутосомно-доминантный |
| Тип 3 (WS3) | 148820 | PAX3 | 2q36.1 | Аутосомно-доминантный или аутосомно-рецессивный |
| Тип 4A (WS4A) | 277580 | ЕДНРБ | 13q22 | Аутосомно-доминантный или аутосомно-рецессивный |
| Тип 4B (WS4B) | 613265 | EDN3 | 20q13 | Аутосомно-доминантный или аутосомно-рецессивный |
| Тип 4C (WS4C) | 613266 | SOX10 | 22q13.1 | Аутосомно-доминантный |
лечение
В настоящее время нет лечения или лекарства от синдрома Ваарденбурга. Симптомом, который, скорее всего, имеет практическое значение, является глухота, и к ней относятся так же, как к любой другой необратимой глухоте. В отмеченных случаях могут возникнуть косметические проблемы. Другие аномалии (неврологические, структурные, Болезнь Гиршпрунга ), связанные с синдромом, лечат симптоматически.
Эпидемиология
Распространенность всех типов синдрома Ваарденбурга оценивается примерно в 1 случай на 42 000.[5][8] Типы 1 и 2 являются наиболее распространенными, а тип 1 - немного более распространенным.[30][31] В обзоре, проведенном в 2015 году с участием 417 пациентов, было обнаружено, что тип 1 является наиболее распространенным типом, охватывая около половины всех случаев (47%), в то время как тип 2 был вторым по распространенности типом, охватывая около трети (33%). .[8] Подавляющее большинство (около 85%) случаев 2-го типа относятся к типу 2А.[8] Распространенность типа 2B неизвестна, поскольку о нем сообщалось только в одном исследовании 1996 года.[22] Тип 2C пока обнаружен только в одной итальянской семье,[23][24] и тип 2D был обнаружен только у 2 неродственных пациентов по состоянию на 2018 год.[Обновить].[13][8][32] Количество известных случаев типа 2E, связанных с неврологическими отклонениями, составило 23 по состоянию на 2017 год.[Обновить],[33] а количество остальных неизвестно. Тип 3 встречается реже, чем типы 1, 2 и 4,[34] составляющих менее 2% случаев.[8] На тип 4 приходится около пятой части случаев (19%). Из его подтипов тип 4C является наиболее распространенным (около 71% от типа 4), за ним следуют тип 4A (19%) и тип 4B (10%).[8]
Подсчитано, что синдром Ваарденбурга присутствует у 2–5% врожденно глухих людей. Врожденная глухота составляет около половины глухоты в целом.[8] Примерно 1 из 30 учеников школ для глухих страдает синдромом Ваарденбурга. Различная форма проявления синдрома затрудняет получение точных данных о его распространенности.[8]
История
Ранние описания
В 1916 году голландский офтальмолог Ян ван дер Хов (1878–1952) описал пару девочек-близнецов с глухотой и особым типом расстройства слуха. блефарофимоз, считается антиутопией canthorum, обнаруживаемой при синдроме Ваарденбурга типов 1 и 3.[8][35] Блефарофимоз - это веки, которые недоразвиты так, что они постоянно закрывают часть глаза.
В 1926 году немецкий врач Ирмгард Менде описал семью из четырех поколений, в которой пятеро детей имели симптомы депигментации волос, кожи и глаз, глухоты и "монголоид «внешний вид (позднее Ваарденбург приписал это описание антиутопии canthorum).[36][35] Позже это привело к тому, что в некоторых базах данных был зарегистрирован синоним «синдром Менде».[11][37]
В 1929 году голландский врач К. Т. А. Хальбертсма описал семейную модель dystopia canthorum,[38][35] а в 1930 году итальянский врач Винченцо Гуальди[39] (1891–1976) также подтвердили наследственность dystopia canthorum.[36] Позднее это привело к тому, что в некоторых базах данных был зарегистрирован синоним синдрома Ван дер Хув-Хальбертсма-Ваарденбург-Гуальди.[11]
В 1947 году швейцарский офтамолог Дэвид Кляйн (1908–1993) впервые сообщили о пациенте с двусторонней глухотой, дефицитом пигментации, характерными чертами лица и уродством рук. Хотя это было первое полное описание пациента с синдромом Ваарденбурга 3-го типа, современные клиницисты не считают описанный им синдром таким же, как описанный Ваарденбургом четыре года спустя, отчасти из-за того, насколько серьезными были пороки развития руки в его теле. терпеливый.[40]
Синдром был впервые полностью формализован и описан голландским офтальмологом и генетиком. Петрус Йоханнес Ваарденбург (1886–1979) в 1951 г.[9][10] Описанное им состояние теперь классифицируется как синдром Ваарденбурга типа 1.
Описание подтипов
Тип 2 был впервые установлен в 1971 году, когда в ходе исследования было обнаружено, что у некоторых пациентов с синдромом Ваарденбурга не было канторной дистопии.[7][41] Исследование 1977 года подтвердило семейный образец этого другого представления.[7] Два исследования 1994 г. впервые подтвердили связь между этим типом синдрома Ваарденбурга и мутациями в MITF ген (теперь классифицируется как тип 2A), расположенный на хромосоме 3 в локусе 3p14.1 – p12.3.[7]
Тип 2B был впервые установлен в 1994 году, когда то же исследование, которое обнаружило мутации в MITF у пациентов с синдромом Ваарденбурга 2 типа также обнаружено, что у некоторых пациентов не было никаких мутаций в этой области.[21][42] Второе исследование 1994 г. обнаружило связь с хромосомой 1 в локусе 1p21 – p13.3. Это стало известно как тип 2B состояния (с обозначенным геном WS2B ), однако с тех пор это не было задокументировано, и ответственный ген остается неизвестным.[21][22]
Тип 2C был установлен в 2001 году, когда исследование итальянской семьи с признаками синдрома Ваарденбурга типа 2 показало, что они были вызваны неизвестным геном на хромосоме 8 в локусе 8q23, который был нарушен хромосомная транслокация. В ходе исследования было установлено предварительное название гена, WS2C. Однако с тех пор мутации в этой области у пациентов с синдромом Ваарденбурга не обнаруживались.[23][24]
Тип 2D был создан в 2002 году в ходе исследования, направленного на поиск мутаций в человеческой версии SNAI2 ген, который, как известно, вызывает депигментацию у мышей, обнаружил делеции обеих копий этого гена у 2 неродственных индивидуумов с синдромом Ваарденбурга типа 2. С тех пор мутации в обеих копиях этого гена не были обнаружены у людей с синдромом Ваарденбурга 2 типа.[8]
Тип 2E был впервые установлен в 1996 году, когда в ходе исследования была выявлена девочка с симптомами синдрома Ваарденбурга типа 2, но с дополнительным недоразвитием перед глазами, приводящее к слепоте. В 1999 году было обнаружено, что у нее есть мутация. SOX10 ген, а более поздние исследования подтвердили связь между мутациями в этом гене и этим фенотипом, а также неврологические симптомы, такие как задержка развития.[4]
Тип 3 впервые получил свое название от Гудмана. и другие. в 1981 году в сотрудничестве с Klein, в котором они установили связь с аномалиями рук, о которых впервые сообщил Klein в 1947 году.[40] Мутации в PAX3 были впервые связаны с этим фенотипом в 1992 году.[2]
Коморбидность с Болезнь Гиршпрунга, который позже будет составлять тип 4, впервые был замечен в различных исследованиях в 1970-х годах. Индийский педиатр Кришнакумар Шах и его коллеги впервые обозначили этот синдром как возможный вариант синдрома Ваарденбурга в 1981 году.[43] Вариант был впервые приписан мутации в ЕДНРБ в 1994 г. (теперь классифицируется как тип 4А).[3] Тип 4B был установлен в 1996 году, когда мутации в EDN3 было обнаружено, что они приводят к этому типу синдрома Ваарденбурга,[27] и тип 4C был впервые установлен в 1998 году, когда мутации в SOX10 также было обнаружено, что они приводят к этому типу.[16]
Общество и культура
Популярная культура
- Роман 2001 года Шок от Робин Кук упоминает персонажа с расстройством.[44]
- Энцо Маклауд, главный герой Питер Мэй серия книг 2006–2017 гг. Файлы Энцо, страдает синдромом Ваарденбурга. У него глаза разного цвета, а в волосах есть белая полоска.[45][46]
- В 2011 году 6 сезон эпизод из Кости «Знаки в тишине», команда должна раскрыть дело, в котором у подозреваемого в убийстве синдром Ваарденбурга.[47]
- Книга 2013 года Реконструкция Амелии от Кимберли МакКрайт есть несколько персонажей с симптомами Ваарденбурга.[48]
- Книга 2014 года Ближе, чем вы думаете от Карен Роуз имеет трех персонажей, братьев и сестер, с синдромом Ваарденбурга.[49]
- Книга 2017 года Убийство в храме майя М.Дж.Мандрагора показывает несколько персонажей с синдромом Ваарденбурга.[50][нужен лучший источник ]
- Роман 2019 года Сеть Whisper Чендлер Бейкер использует синдром в качестве сюжета.[нужна цитата ]
Известные люди
- Популярный канадский видеоблогер на YouTube Стеф Санджати имеет синдром Ваарденбурга 1 типа.[51]
Другие животные

Синдром Ваарденбурга типа 2А (с мутацией в MITF) был обнаружен у собак, Крупный рогатый скот Fleckvieh, норки, мыши и золотой хомяк.[52] Дегенерация улитки и мешочек, как видно из синдрома Ваарденбурга, также был обнаружен у глухих белых кошек, Далматинцы и других пород собак, белых норок и мышей.[53]
Домашние кошки с голубыми глазами и белой шерстью часто бывают полностью глухими.[54] Глухота гораздо чаще встречается у белых кошек, чем у кошек других окрасов. Согласно ASPCA Полное руководство по кошкам », от 17 до 20 процентов белых кошек с не голубыми глазами являются глухими; 40 процентов« разноглазых »белых кошек с одним голубым глазом глухие; и от 65 до 85 процентов голубоглазых белых кошек глухие. глухой."[55] Хотя было проведено мало исследований, чтобы связать это с генами, которые, как известно, участвуют в синдроме Ваарденбурга у человека, генетическое нарушение развития нервного гребня может привести к этому проявлению и у кошек.[56] Один из генов, который приводит к глухоте и белой шерсти у кошек при мутации, КОМПЛЕКТ,[57] было обнаружено увеличение MITF выражение.[58]
Смертельный белый синдром синдром у лошадей, вызванный мутациями в обеих копиях ЕДНРБ. Это приводит к смерти от псевдообструкции кишечника из-за болезни Гиршпрунга. Мутация в единственном экземпляре ЕДНРБоднако, как и при синдроме Ваарденбурга типа 4A, образуются пятнистые белые пятна. оверо пальто при глухоте.[59]
Хорьки с синдромом Ваарденбурга есть небольшая белая полоса вдоль макушки или затылка, а иногда и вниз по шее (известная как «пылающий» рисунок шерсти), или сплошная белая голова от носа до плеч (известная как «панда» "узор пальто). У пораженных хорьков череп часто немного более плоский, а глаза широко посажены, чем у здоровых хорьков. Поскольку у здоровых хорьков плохой слух, глухоту можно определить только по отсутствию реакции на громкие звуки. Поскольку это наследственное заболевание, пораженных животных нельзя использовать для разведения. Исследование корреляции между вариациями шерсти и глухотой у европейских хорьков показало, что «все (n = 27) панда, американская панда и огненные хорьки были глухими».[60]
Смотрите также
- Синдром Чедиака – Хигаши, подобный синдром, включая иммунодефицит и периферическую невропатию
- Смертельный белый синдром, летальная форма синдрома Ваарденбурга типа 4А у лошадей (вызванная мутациями в обеих копиях ЕДНРБ)
- Синдром Тиц, состояние, сходное с синдромом Ваарденбурга типа 2, включая однородный альбинизм (вызванное мутациями в MITF)
- Болезнь Фогта – Коянаги – Харада, аутоиммунное заболевание, вызывающее увеит, пятнистую депигментацию и симптомы внутреннего уха
использованная литература
- ^ а б c d «Запись OMIM - # 193500 - ВАРДЕНБУРГСКИЙ СИНДРОМ, ТИП 1; WS1». omim.org. Получено 2019-12-07.
- ^ а б c d е «Запись OMIM - № 148820 - ВАРДЕНБУРГСКИЙ СИНДРОМ, ТИП 3; WS3». omim.org. Получено 2019-12-07.
- ^ а б c d «Запись OMIM - № 277580 - ВАРДЕНБУРГСКИЙ СИНДРОМ, ТИП 4A; WS4A». omim.org. Получено 2019-12-07.
- ^ а б c d «Запись OMIM - № 611584 - ВАРДЕНБУРГСКИЙ СИНДРОМ, ТИП 2E; WS2E». omim.org. Получено 2019-12-07.
- ^ а б c d е ж г час Пинго, Вероника; Энте, Доротея; Моал, Флоренс Дасто-Ле; Гуссенс, Мишель; Марлин, Сандрин; Бондюран, Надеж (2010). «Обзор и обновление мутаций, вызывающих синдром Ваарденбурга». Человеческая мутация. 31 (4): 391–406. Дои:10.1002 / humu.21211. ISSN 1098-1004. PMID 20127975. S2CID 12278025.
- ^ а б «Развитие нервного гребня - эмбриология». embryology.med.unsw.edu.au. Получено 2019-12-13.
- ^ а б c d е ж г час «Запись OMIM - № 193510 - ВАРДЕНБУРГСКИЙ СИНДРОМ, ТИП 2A; WS2A». omim.org. Получено 2019-12-07.
- ^ а б c d е ж г час я j k л м п Song, J .; Feng, Y .; Acke, F. R .; Coucke, P .; Vleminckx, K .; Дхоге, И. Дж. (2016). «Потеря слуха при синдроме Ваарденбурга: систематический обзор». Клиническая генетика. 89 (4): 416–425. Дои:10.1111 / cge.12631. ISSN 1399-0004. PMID 26100139. S2CID 23834634.
- ^ а б Чандра Мохан, Сетти. Л. Н. (01.09.2018). «Случай синдрома Ваарденбург-Шаха в семье с обзором литературы». Журнал отологии. 13 (3): 105–110. Дои:10.1016 / j.joto.2018.05.005. ISSN 1672-2930. ЧВК 6291636. PMID 30559775.
- ^ а б Ваарденбург PJ (сентябрь 1951). «Новый синдром, сочетающий аномалии развития век, бровей и корня носа с пигментными аномалиями радужной оболочки глаза и волос на голове и с врожденной глухотой; Dystopia canthi medialis et punctorum lacrimalium lateoversa, Hyperplasia supercilii medialis et radicis nasi, Heterochromia iridum totalis sive partialis, Albinismus Circriptus (лейцизм, полиоз) и Surditas congenita (surdimutitas)". Am. J. Hum. Genet. 3 (3): 195–253. ЧВК 1716407. PMID 14902764.
- ^ а б c «Синдром Ваарденбурга | Информационный центр по генетическим и редким заболеваниям (GARD) - программа NCATS». rarediseases.info.nih.gov. Получено 2018-04-17.
- ^ «Синдром Ваарденбурга». Домашний справочник по генетике. Октябрь 2012 г.
- ^ а б «Запись OMIM - № 608890 - ВАРДЕНБУРГСКИЙ СИНДРОМ, ТИП 2D; WS2D». omim.org. Получено 2019-12-07.
- ^ Барал В., Чауи А., Ватанабе И., Гуссенс М., Атти-Битах Т., Марлин С., Пинго В., Бондюран Н. (2012). «Скрининг регуляторных регионов MITF и SOX10 при синдроме Ваарденбурга 2 типа». PLOS ONE. 7 (7): e41927. Bibcode:2012PLoSO ... 741927B. Дои:10.1371 / journal.pone.0041927. ЧВК 3407046. PMID 22848661.
- ^ а б Pierpont, J. W .; St Jacques, D .; Seaver, L.H .; Эриксон, Р. П. (март 1995 г.). «Семья с необычным синдромом Ваарденбурга типа I (WSI), расщелиной губы (неба) и болезнью Гиршпрунга не связана с PAX 3». Клиническая генетика. 47 (3): 139–143. Дои:10.1111 / j.1399-0004.1995.tb03946.x. ISSN 0009-9163. PMID 7634536. S2CID 24530898.
- ^ а б c «Запись OMIM - № 613266 - ВАРДЕНБУРГСКИЙ СИНДРОМ, ТИП 4C; WS4C». omim.org. Получено 2019-12-07.
- ^ «Сирота: поиски болезни». www.orpha.net. Получено 2019-12-10.
- ^ Verheij, Johanna B.G.M .; Сивал, Дебора А .; van der Hoeven, Johannes H .; Vos, Yvonne J .; Meiners, Linda C .; Brouwer, Oebele F .; ван Эссен, Энтони Дж. (январь 2006 г.). «Синдром Шах-Ваарденбурга и PCWH, связанный с мутациями SOX10: отчет о болезни и обзор литературы». Европейский журнал детской неврологии. 10 (1): 11–17. Дои:10.1016 / j.ejpn.2005.10.004. ISSN 1090-3798. PMID 16504559.
- ^ Williams, Antionette L .; Бонсак, Бренда Л. (июнь 2015 г.). "Производные нервного гребня в развитии глаз: проницательный глаз бури". Исследование врожденных пороков, часть C: эмбрион сегодня: обзоры. 105 (2): 87–95. Дои:10.1002 / bdrc.21095. ISSN 1542-975X. ЧВК 5262495. PMID 26043871.
- ^ Каваками, Акинори; Фишер, Дэвид Э. (июнь 2017 г.). «Главная роль фактора транскрипции, связанного с микрофтальмией, в биологии меланоцитов и меланомы». Лабораторные исследования; Журнал технических методов и патологии. 97 (6): 649–656. Дои:10.1038 / labinvest.2017.9. ISSN 1530-0307. PMID 28263292.
- ^ а б c «Запись OMIM -% 600193 - ВАРДЕНБУРГСКИЙ СИНДРОМ, ТИП 2B; WS2B». www.omim.org. Получено 2019-12-23.
- ^ а б c Lalwani, A.K .; Сан-Агустин, Т. Б.; Уилкокс, Э. Р. (1994-09-01). «Локус синдрома Ваарденбурга типа II отображается на хромосоме 1p13.3-2.1». Американский журнал генетики человека. 55 (Приложение 3). OSTI 133315.
- ^ а б c «Запись OMIM -% 606662 - ВАРДЕНБУРГСКИЙ СИНДРОМ, ТИП 2C; WS2C». omim.org. Получено 2019-12-07.
- ^ а б c Селикорни, Анджело; Гернери, Сильвана; Ратти, Антония; Пиццути, Антонио (январь 2002 г.). «Цитогенетическое картирование нового локуса для синдрома Ваарденбурга II типа». Генетика человека. 110 (1): 64–67. Дои:10.1007 / s00439-001-0643-9. ISSN 0340-6717. PMID 11810298. S2CID 24411957.
- ^ Санчес-Мартин, Мануэль; Перес-Лосада, Хесус; Родригес-Гарсия, Аранча; Гонсалес-Санчес, Белен; Корф, Брюс Р .; Kuster, W .; Мосс, Селия; Spritz, Ричард А .; Санчес-Гарсия, И. (2003). «Делеция гена SLUG (SNAI2) приводит к пегому поведению человека». Американский журнал медицинской генетики, часть A. 122A (2): 125–132. Дои:10.1002 / ajmg.a.20345. ISSN 1552-4833. PMID 12955764. S2CID 33811699.
- ^ Текин, М .; Bodurtha, J. N .; Nance, W. E .; Пандья, А. (2001). «Синдром Ваарденбурга типа 3 (синдром Кляйна – Ваарденбурга), сегрегационный с гетерозиготной делецией в парном бокс-домене PAX3: простой вариант или истинный синдром?». Клиническая генетика. 60 (4): 301–304. Дои:10.1034 / j.1399-0004.2001.600408.x. ISSN 1399-0004. PMID 11683776. S2CID 24025063.
- ^ а б «Запись OMIM - № 613265 - ВАРДЕНБУРГСКИЙ СИНДРОМ, ТИП 4B; WS4B». omim.org. Получено 2019-12-07.
- ^ Ян Т., Ли Х, Хуан Ц., Ли Л., Чай И, Сунь Л., Ван Х, Чжу И, Ван З, Хуанг З, Ли И, Ву Х (январь 2013 г.). «Двойные гетерозиготные мутации MITF и PAX3 приводят к синдрому Ваарденбурга с повышенной пенетрантностью в пигментных дефектах». Clin. Genet. 83 (1): 78–82. Дои:10.1111 / j.1399-0004.2012.01853.x. PMID 22320238. S2CID 34173541.
- ^ Цукамото К., Накамура Ю., Ниикава Н. (март 1994 г.). «Выделение двух изоформ транскриптов гена PAX3 и их тканеспецифическая альтернативная экспрессия в тканях взрослого человека». Гм. Genet. 93 (3): 270–4. Дои:10.1007 / bf00212021. PMID 7545913. S2CID 36749688.
- ^ «Орфанет: синдром Ваарденбурга типа 1». www.orpha.net. Получено 2019-12-10.
- ^ «Синдром Ваарденбурга II типа» (PDF). Orphanet. 2005. Получено 10 декабря 2019.
- ^ Салим, Мохаммед Д. (2019). «Биология развития меланоцитов человека, пибальдизм и синдром Ваарденбурга». Детская дерматология. 36 (1): 72–84. Дои:10.1111 / pde.13713. ISSN 1525-1470. PMID 30561083.
- ^ Богданова ‐ Михайлова, Петя; Александр, Майкл Д .; Мерфи, Раймонд П. Дж .; Мерфи, Шинеад М. (2017). «Синдром Ваарденбурга: редкая причина наследственной невропатии из-за мутации SOX10». Журнал периферической нервной системы. 22 (3): 219–223. Дои:10.1111 / jns.12221. ISSN 1529-8027. PMID 28544110. S2CID 13676694.
- ^ "Орфанет: синдром Ваарденбурга тип 3". www.orpha.net. Получено 2019-12-10.
- ^ а б c De Haas, E. B.H .; Тан, К. Э. У. П. (1966-01-01). «Синдром Ваарденбурга». Documenta Ophthalmologica. 21 (1): 239–282. Дои:10.1007 / BF00184136. ISSN 1573-2622. S2CID 10163905.
Ваарденбург (1951, 1957, 1961) выразил убеждение, что все эти случаи неосложненного блефарофимоза на самом деле относятся к его синдрому и что этот тип dystopia canthorum не встречается как отдельный признак. ... Что касается случаев Mende, [Waardenburg] считает, что «монголоидный компонент» у этих пациентов на самом деле был вызван dystopia canthorum.
- ^ а б Кляйн, Д. (февраль 1983 г.). «Историческая справка и доказательства доминантного наследования синдрома Клейна-Ваарденбурга (тип III)». Американский журнал медицинской генетики. 14 (2): 231–239. Дои:10.1002 / ajmg.1320140205. ISSN 0148-7299. PMID 6340503.
- ^ Стедман, Томас Латроп (2005). Медицинские эпонимы Стедмана. Липпинкотт Уильямс и Уилкинс. ISBN 978-0-7817-5443-9.
- ^ "Over twee op elkaar gelijkende, in wezen echter verschillende aangeboren oogafwijkingen". Nederlands Tijdschrift для Geneeskunde (на голландском). 2009-12-02. Получено 2019-12-10.
- ^ Annuario del Ministero dell'Educazione nazionale (на итальянском). Provveditorato generale dello Stato. 1935. с. 142.
- ^ а б Goodman, R.M .; Lewithal, I .; Соломон, А .; Кляйн, Д. (апрель 1982 г.). «Поражение верхних конечностей при синдроме Клейна-Ваарденбурга». Американский журнал медицинской генетики. 11 (4): 425–433. Дои:10.1002 / ajmg.1320110407. ISSN 0148-7299. PMID 7091186.
- ^ Ариас, С. (март 1971 г.). «Генетическая гетерогенность в синдроме Ваарденбурга». Врожденные дефекты. Серия оригинальных статей. 07 (4): 87–101. ISSN 0547-6844. PMID 5006208.
- ^ Hughes, A.E .; Ньютон, В. Э .; Лю, X. Z .; Рид, А. П. (август 1994 г.). «Ген синдрома Ваарденбурга типа 2 картирован близко к человеческому гомологу гена микрофтальмии на хромосоме 3p12-p14.1». Природа Генетика. 7 (4): 509–512. Дои:10.1038 / ng0894-509. ISSN 1061-4036. PMID 7951321. S2CID 2913481.
- ^ Shah, Krishnakumar N .; Dalal, Subhash J .; Desai, Meena P .; Sheth, Prem N .; Joshi, Nana C .; Амбани, Лалит М. (1981-09-01). «Белый чуб, нарушение пигментации радужной оболочки и болезнь Гиршпрунга длинного сегмента: возможный вариант синдрома Ваарденбурга». Журнал педиатрии. 99 (3): 432–435. Дои:10.1016 / S0022-3476 (81) 80339-3. ISSN 0022-3476. PMID 7264803.
- ^ Кук, Робин (12 декабря 2011 г.). Шок. Пан Макмиллан. п. 175. ISBN 978-1-4472-1796-1.
- ^ Май, Питер (30.05.2013). Необычные люди: Энцо Маклеод 1. Quercus. п. 32. ISBN 978-1-78206-885-3.
- ^ Май, Питер (13.06.2013). Blacklight Blue: Энцо Маклеод 3. Quercus. п. 76. ISBN 978-1-78206-887-7.
- ^ "Bones Recap 6.21" Знаки в безмолвии "- Журнал Persephone". Получено 2019-12-14.
- ^ МакКрайт, Кимберли (1 апреля 2013 г.). Реконструкция Амелии. Саймон и Шустер. ISBN 978-1-4711-2944-5.
- ^ Роза, Карен (2014-11-06). Ближе, чем вы думаете (Серия Цинциннати, книга 1). Заголовок. ISBN 978-0-7553-8999-5.
- ^ минони. "Знакомьтесь, хороший сыщик". Amazon.com. Получено 2019-12-14.
- ^ Эдвардс, Люси (2018). "'Я понимаю, ты трансгендер, но что с твоим лицом?'". Новости BBC. Получено 2018-01-28.
- ^ МАРКАКИС, МАРИОС Н .; SOEDRING, VIBEKE E .; ДАНТЦЕР, ВИБЕКЕ; КРИСТЕНСЕН, КНУД; АНИСТОРОАЭЙ, РАЗВАН (01.08.2014). «Связь гена MITF с фенотипом слуха и пигментации у белой американской норки Хедлунда (Neovison vison)». Журнал генетики. 93 (2): 477–481. Дои:10.1007 / s12041-014-0370-3. HDL:10067/1211550151162165141. ISSN 0973-7731. PMID 25189243. S2CID 16725018.
- ^ Штамм, Джордж М. (2015). «Генетика глухоты у домашних животных». Границы ветеринарии. 2: 29. Дои:10.3389 / fvets.2015.00029. ISSN 2297-1769. ЧВК 4672198. PMID 26664958.
- ^ Уэбб А.А., Каллен С.Л. (июнь 2010 г.). «Неврологические и нейроофтальмологические заболевания, связанные с окрасом и структурой окраса шерсти». Мочь. Вет. J. 51 (6): 653–7. ЧВК 2871368. PMID 20808581.
- ^ Ричардс Дж. (1999). Полное руководство ASPCA для кошек: все, что вам нужно знать о выборе и уходе за своим питомцем. Книги хроники. п. 71. ISBN 9780811819299.
- ^ Omenn, Gilbert S .; McKusick, Victor A .; Горлин, Роберт Дж. (1979). «Ассоциация синдрома Ваарденбурга и мегаколона Хиршпрунга». Американский журнал медицинской генетики. 3 (3): 217–223. Дои:10.1002 / ajmg.1320030302. ISSN 1096-8628. PMID 484594.
Глухой, голубоглазый белый кот, отмеченный Бри [1829] и Дарвином [1892] и изученный гистологически на рубеже веков [Александр, 1900], имеет изменчивую клиническую и гистологическую картину из-за любого из двух аутосомно-доминантные гены ... Эти плейотропные эффекты отдельных генов могут быть объяснены воздействием на клетки нервного гребня, участвующие в происхождении всех тканей, пораженных синдромом Ваарденбурга [Weston, 1969].
- ^ Дэвид, Виктор А .; Менотти-Раймонд, Мэрилин; Уоллес, Андреа Кутс; Рулке, Мелодия; Келер, Джеймс; Leighty, Роберт; Эйзирик, Эдуардо; Ханна, Стивен С .; Нельсон, Джордж; Schäffer, Alejandro A .; Коннелли, Кэтрин Дж. (2014-10-01). «Включение эндогенного ретровируса в онкоген KIT определяет белые и белые пятна у домашних кошек». G3: Гены, геномы, генетика. 4 (10): 1881–1891. Дои:10.1534 / g3.114.013425. ISSN 2160-1836. ЧВК 4199695. PMID 25085922.
- ^ Ли, Юль-Нам; Брандаль, Стефани; Ноэль, Пьер; Венцель, Эрик; Mendell, Joshua T .; Макдевитт, Майкл А .; Капур, Рувим; Картер, Мелоди; Меткалф, Дин Д .; Такемото, Клиффорд М. (31 марта 2011 г.). «Передача сигналов KIT регулирует экспрессию MITF через miRNA при нормальной и злокачественной пролиферации тучных клеток». Кровь. 117 (13): 3629–3640. Дои:10.1182 / blood-2010-07-293548. ISSN 0006-4971. ЧВК 3072881. PMID 21273305.
- ^ Магдезиан, К. Гэри; Уильямс, Д. Колетт; Алеман, Моника; LeCouteur, Ричард А .; Мэдиган, Джон Э. (15 ноября 2009 г.). «Оценка глухоты у американских пейнтбольных лошадей по фенотипу, стволовым слуховым ответам и генотипу рецептора B эндотелина». Журнал Американской ветеринарной медицинской ассоциации. 235 (10): 1204–1211. Дои:10.2460 / javma.235.10.1204. ISSN 0003-1488. PMID 19912043.
- ^ Piazza S, Abitbol M, Gnirs K, Huynh M, Cauzinille L (май 2014 г.). «Распространенность глухоты и ассоциации с вариациями шерсти у принадлежащих клиенту хорьков». Варенье. Вет. Med. Assoc. 244 (9): 1047–52. Дои:10.2460 / javma.244.9.1047. PMID 24739114.
внешние ссылки
| Классификация | |
|---|---|
| Внешние ресурсы |
- GeneReviews / NCBI / NIH / UW запись о синдроме Ваарденбурга типа I
- Каталог генетических заболеваний OMIM - синдром Ваарденбурга