Иммуномодулирующий имидный препарат - Immunomodulatory imide drug - Wikipedia
| Иммуномодулирующий имидный препарат | |
|---|---|
| Класс препарата | |
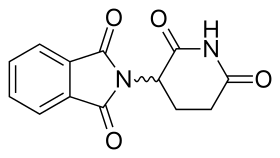 Талидомид | |
| Идентификаторы класса | |
| Использовать | Узловатая лепрозная эритема, множественная миелома, миелодиспластический синдром, острый миелоидный лейкоз и другие иммунологические состояния |
| Код УВД | L04AX |
| Биологическая мишень | TNF, Ил-6, VEGF, NF-kB, так далее. |
| Клинические данные | |
| Drugs.com | Классы наркотиков |
| В Викиданных | |
Иммуномодулирующие имидные препараты (IMiDs) являются классом иммуномодулирующие препараты[1] (препараты, регулирующие иммунные ответы ) содержащий имид группа. Класс IMiD включает талидомид и его аналоги (леналидомид, помалидомид, ибердомид и апремиласт ).[1] Эти препараты могут также называться «модуляторами цереблона». Цереблон белок, на который нацелен этот класс препаратов.
Название "IMiD" намекает как к «IMD» для «иммуномодулирующего препарата», так и к формам имид, имидо-, имид-, и имид.
Разработка аналогов талидомида была ускорена открытием антиангиогенный и противовоспалительное средство свойства препарата открывают новый способ борьбы с раком, а также с некоторыми воспалительными заболеваниями после того, как он был запрещен в 1961 году. В том числе проблемы с талидомидом; тератогенные побочные эффекты, высокая частота других побочных реакций, плохая растворимость в воде и плохое всасывание из кишечника.
В 1998 году талидомид был одобрен США. Управление по контролю за продуктами и лекарствами (FDA) для использования у впервые диагностированных множественная миелома (MM) в соответствии со строгими правилами.[2] Это привело к развитию ряда аналоги с меньшим количеством побочные эффекты и увеличился потенция который включает в себя леналидомид, помалидомид и апремиласт, все из которых в настоящее время продаются и производятся Celgene.
Поколения
Существует три поколения IMiD, каждое последующее поколение лучше переносится и более активно против воспалительных и злокачественных заболеваний.[1]
- Первое поколение - талидомид
- Второе поколение - леналидомид и помалидомид
- Третье поколение - апремиласт
История
Талидомид был первоначально выпущен в Федеральная Республика Германии (Западная Германия) под маркой Contergan 1 октября 1957 г. Chemie Grünenthal (сейчас же Grünenthal ). Препарат в основном назначали как успокаивающее или снотворное, но его также использовали как противорвотное средство (утреннее недомогание у беременных) и успокаивающее. Препарат был запрещен в 1961 г. тератогенный свойства не наблюдались. Проблемы с талидомидом были, помимо тератогенных побочных эффектов, высокой частотой других неблагоприятные реакции вместе с бедными растворимость в воде и поглощение от кишечник.[3][4] Побочные реакции включают: периферическая невропатия у подавляющего большинства пациентов, запор, тромбоэмболия вместе с дерматологический осложнения.[5]
Через четыре года после того, как талидомид был изъят с рынка из-за его способности вызывать серьезные врожденные дефекты, его противовоспалительные свойства были обнаружены, когда пациенты, страдающие от узловатая лепрозная эритема (ЭНЛ) использовали талидомид в качестве седативного средства, и он уменьшил как клинические признаки, так и симптомы болезни. Было обнаружено, что талидомид ингибирует фактор некроза опухоли альфа (TNF-α) в 1991 г. (5a Sampaio, Sarno, Galilly Cohn and Kaplan, JEM 173 (3) 699-703, 1991). TNF-α представляет собой цитокин произведено макрофаги иммунной системы, а также медиатор воспалительного ответа. Таким образом, препарат эффективен против некоторых воспалительных заболеваний, таких как ENL (6a Sampaio, Kaplan, Miranda, Nery ..... JID 168 (2) 408-414 2008). В 1994 году было обнаружено, что талидомид обладает антиангиогенной активностью.[6] и противоопухолевая активность[7] которые послужили толчком к началу клинических испытаний рака, включая множественную миелому. Открытие противовоспалительной, антиангиогенной и противоопухолевой активности талидомида повысило интерес к дальнейшим исследованиям и синтез более безопасных аналогов.[8][9]
Леналидомид - это первый аналог талидомида, который поступил на рынок. Он значительно более эффективен, чем его исходное лекарство, только с двумя различиями на молекулярном уровне, с добавлением аминогруппа в положении 4 фталоильного кольца и удаление карбонил группа из фталоильного кольца.[10]Разработка леналидомида началась в конце 1990-х годов, а клинические испытания леналидомида начались в 2000 году. В октябре 2001 года леналидомид получил статус орфанных для лечения ММ. В середине 2002 г. он перешел в фазу II, а к началу 2003 г. - в фазу III. В феврале 2003 г. FDA предоставило леналидомиду ускоренный статус для лечения рецидивирующего или рефрактерного ММ.[8]В 2006 г. он был одобрен для лечения ММ вместе с дексаметазоном, а в 2007 г. Европейское агентство по лекарствам (EMA). В 2008 г. в ходе исследования фазы II была выявлена эффективность лечения Неходжкинской лимфомы.[11]
Помалидомид (3-аминоталидомид) был вторым аналогом талидомида, поступившим в клинику, будучи более эффективным, чем оба его предшественника.[12] Впервые сообщалось в 2001 г., что помалидомид напрямую ингибирует пролиферацию миеломных клеток и, таким образом, ингибирует ММ как в опухолевых, так и в сосудистых отделах.[13] Эта двойная активность помалидомида делает его более эффективным, чем оба талидомида. in vitro и in vivo.[14] Этот эффект не связан с ингибированием TNF-α, поскольку сильнодействующие ингибиторы TNF-α, такие как ролипрам и пентоксифиллин, не ингибируют ни рост миеломных клеток, ни ангиогенез.[9] Сообщалось о повышении регуляции интерферона гамма, ИЛ-2 и ИЛ-10 для помалидомида, что может способствовать его антиангиогенной и противомиеломной активности.
Разработка
Молекула талидомида является синтетическим производным глютаминовая кислота и состоит из глутаримидного кольца и фталоильного кольца (рис. 5).[15][16] Его ИЮПАК название 2- (2,6-диоксопиперидин-3-ил) изоиндол-1,3-дион и имеет один хиральный центр[15]После того, как было сообщено о селективном ингибировании талидомидом TNF-α, были предприняты новые усилия по клинической разработке талидомида. Клиническая разработка привела к открытию новых аналогов, направленных на улучшение активности и уменьшение побочных эффектов.[8][17]
Клинически талидомид всегда использовался как рацемат. Обычно S-изомер связан с печально известным тератогенным действием талидомида и р-изомер не обладает тератогенными свойствами, но обладает седативным действием,[8] однако эта точка зрения широко обсуждается, и утверждается, что модель на животных, что эти разные р- и S- эффекты наблюдались в нечувствительности к тератогенным эффектам талидомида. Более поздние сообщения о кроликах, которые являются чувствительным видом, выявили тератогенные эффекты обоих изомеров.[8][15][16][17] Более того, энантиомеры талидомида оказались взаимопереход in vivo из-за кислого хирального водорода в асимметричном центре (показан для аналога EM-12 на рисунке 3),[16][17] так что план администрирования очищенного сингла энантиомер избежать тератогенного воздействия, скорее всего, будет напрасно.[8][15][16]
Разработка леналидомида и помалидомида

Один из интересующих аналогов представляет собой изоиндолиноновое замещение фталоильного кольца. Ему было присвоено название ЭМ-12 (рисунок 3). Считалось, что эта замена увеличит биодоступность вещества из-за повышенной стабильности. Сообщалось, что эта молекула является даже более сильным тератогенным агентом, чем талидомид, для крыс, кроликов и обезьян. Кроме того, эти аналоги являются более сильными ингибиторами ангиогенеза, чем талидомид.[13] Кроме того, амино-талидомид и амино-ЕМ-12 были мощными ингибиторами TNF-α.[16] Эти два аналога позже получили название леналидомид, который представляет собой амино аналог ЕМ-12, и помалидомид, амино аналог талидомида.[8]
Разработка апремиласта

После обнаружения нового набора аналогов талидомида, а именно 3- (1,3-диоксо-1,3-дигидроизоиндол-2-ил) -3- (3,4-диметоксифенил) пропионовой кислоты (не показана), которые ингибировали PDE4 деятельность началась работа по оптимизации деятельности. Для этого исследователи использовали известную структуру часть, 3,4-диалкоксифенил, который является признанным фармакофор в ингибиторах PDE4, таких как ролипрам (Рисунок 6) и рофлумиласт и добавил его в структуру ранее упомянутой аналоговой серии. После настройки структуры и тестирования различных замен в положении 4 фталоильного кольца и карбоксильной кислоты исследователи наконец обнаружили молекулу, которая эффективно ингибирует PDE4 и TNF-α, которую они позже назвали апремиластом (рис. 4). В S-энантиомер апремиласта был выбран, поскольку он был более активным энантиомером.[15] Поскольку в структуре апремиласта отсутствует кислый хиральный водород, он не должен рацемизироваться. in vivoв отличие от талидомида, леналидомида и помалидомида.[16][17]
Медицинское использование
В первую очередь IMiD используются в медицине для лечения раки и аутоиммунные заболевания (включая тот, который является ответом на инфекцию проказа ).[18] Показания для этих агентов, получивших одобрение регулирующих органов, включают:[19]
- Миелодиспластический синдром, условие-предшественник острый миелоидный лейкоз
- Узловатая эритема, осложнение проказы
- Множественная миелома
Показания не по назначению, при которых они кажутся многообещающими методами лечения, включают:[20]
- Лимфома Ходжкина
- Амилоидоз, связанный с легкими цепями (AL)
- Начальный миелофиброз (PMF)
- Острый миелоидный лейкоз (AML)
- Рак простаты
- Метастатический карцинома почек (мПКР)
Талидомид
Талидомид был одобрен FDA для ЭНЛ и ММ в сочетании с дексаметазон. EMA также одобрило его для лечения ММ в сочетании с преднизон и / или мелфалан. Признаки сиротства FDA включают: болезнь трансплантат против хозяина, микобактериальная инфекция, рецидивирующая афтозные язвы, тяжелый рецидивирующий афтозный стоматит, первичные злокачественные опухоли головного мозга, связанные со СПИДом тратить синдром, Крона болезнь, Саркома Капоши, миелодиспластический синдром и трансплантация гемопоэтических стволовых клеток.[21][22]
Леналидомид
Леналидомид одобрен почти в 70 странах в комбинации с дексаметазоном для лечения пациентов с ММ, которые ранее получали хотя бы одну терапию. Признаки сиротства включают: диффузная В-клеточная лимфома большого размера, хронический лимфолейкоз и лимфома из клеток мантии.Леналидомид также разрешен к применению при переливании крови. анемия из-за миелодиспластических синдромов низкого или среднего риска, связанных с цитогенетической аномалией делеции 5q с дополнительными цитогенетическими аномалиями или без них, в США, Канаде, Швейцарии, Австралии, Новой Зеландии, Малайзии, Израиле и нескольких странах Латинской Америки, в то время как заявка на получение разрешения на маркетинг в настоящее время оценивается в ряде других стран.[23][24]Многочисленные клинические испытания уже находятся в стадии разработки или проводятся для изучения дальнейшего использования леналидомида, отдельно или в комбинации с другими лекарствами. Некоторые из этих указаний включают: острый миелоидный лейкоз, фолликулярная лимфома, MALT лимфома, Макроглобулинемия Вальденстрема, Красная волчанка, Лимфома Ходжкина, миелодиспластический синдром и др.[25][26]
Помалидомид
Помалидомид был представлен на одобрение FDA 26 апреля 2012 г.[27] 21 июня было объявлено, что препарат пройдет стандартную проверку FDA. Заявление на получение разрешения на продажу было подано в EMA 21 июня 2012 г., и решение может быть принято уже в начале 2013 г. EMA уже присвоило помалидомиду статус сиротства для первичного миелофиброз, ММ, системный склероз, почтовый-полицитемия и постэссенциальный тромбоцитемический миелофиброз.[28]
Апремиласт
По состоянию на сентябрь 2012 г. апремиласт находится в фазе III испытаний для псориаз и фазы II испытаний для ревматоидный артрит. Эффективность в анкилозирующий спондилоартрит тоже проходит испытания.[29] По состоянию на март 2014 г. апремиласт одобрен для лечения псориатического артрита.[30] В сентябре 2014 года FDA США одобрило апремиласт для лечения бляшечного псориаза средней и тяжелой степени.
Побочные эффекты
Основные токсические эффекты одобренных IMiD: периферическая невропатия, тромбоцитопения, анемия и Венозная тромбоэмболия.[20] Может быть повышенный риск вторичных злокачественных новообразований, особенно острый миелоидный лейкоз в тех, кто получает IMiD.[20]
Тератогенность
Тератогенность талидомида была предметом множества споров, и на протяжении многих лет гипотезы Были предложены. Двумя наиболее известными из них были гипотеза антиангиогенеза и гипотеза модели окислительного стресса, со значительными экспериментальными данными, подтверждающими эти две гипотезы относительно тератогенности талидомида.[31]
Недавно появились новые открытия, которые предполагают новый механизм тератогенности. Цереблон 51 kДа белок, локализованный в цитоплазма, ядро и периферическая мембрана клеток во многих частях тела.[32] Он действует как компонент Убиквитинлигаза E3, регулирующие различные процессы развития, в том числе эмбриогенез, канцерогенез и регуляция клеточного цикла через деградацию (убиквитинирование ) неизвестных субстратов. Было показано, что талидомид связывается с церблоном, ингибируя активность убиквитинлигазы E3, что приводит к накоплению субстратов лигазы и подавлению активности фактор роста фибробластов 8 (FGF8) и FGF10. Это нарушает петля положительной обратной связи между двумя факторами роста, что может вызывать как множественные врожденные дефекты, так и противомиеломные эффекты.
Полученные данные также подтверждают гипотезу о том, что увеличение экспрессии церблона является важным элементом противомиеломного эффекта как леналидомида, так и помалидомида.[31] Экспрессия цереблонов была в три раза выше у пациентов, ответивших на лечение, по сравнению с пациентами без ответа, и более высокая экспрессия цереблонов была также связана с частичным или полным ответом, в то время как более низкая экспрессия была связана со стабильным или прогрессирующим заболеванием.[32]
Механизм действия
Механизм их действия не совсем ясен, но известно, что они подавляют выработку фактор некроза опухоли, интерлейкин 6 и иммуноглобулин G и VEGF (что приводит к его антиангиогенным эффектам), костимулирует Т-клетки и NK-клетки и увеличивает интерферон гамма и интерлейкин 2 производство.[33][34][35] Их тератогенные эффекты, по-видимому, опосредованы связыванием с церблон.[36] Апремиласт, с другой стороны, подавляет PDE4.[20]
Считается, что талидомид и его аналоги, леналидомид и помалидомид, действуют аналогичным образом, хотя их точные механизм действия еще полностью не изучен. Считается, что при различных заболеваниях они действуют через разные механизмы. Чистый эффект, вероятно, связан с сочетанием разных механизмов. Однако считается, что апремиласт действует через другой механизм и поэтому будет обсуждаться отдельно. Механизм действия будет объяснен в свете сегодняшних знаний, в основном, в MM (Рисунок 2).
Талидомид, леналидомид и помалидомид
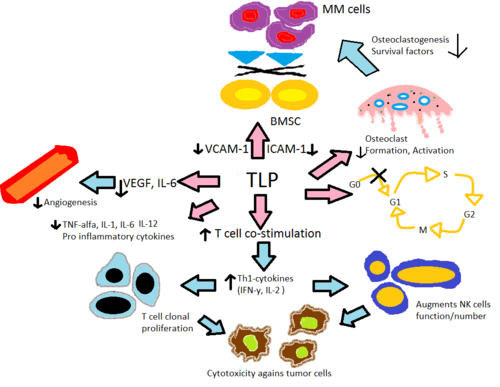
Изменение производства цитокинов
Талидомид и его иммуномодулирующие аналоги изменяют выработку воспалительных цитокинов TNF-α, Ил-1, Ил-6, Ил-12 и противовоспалительный цитокин Ил-10.[32] Считается, что аналоги ингибируют выработку TNF-α, при этом аналоги в 50 000 раз сильнее. in vitro чем исходный препарат талидомид.[37] Считается, что этот механизм связан с усиленной деградацией TNF-α. мРНК, что приводит к уменьшению секретируемого провоспалительного цитокина.[38] Это объясняет эффект талидомида при введении пациентам с ЭНЛ, поскольку у них обычно наблюдается высокий уровень TNF-α в крови и при дерматологических поражениях.[8] В отличие, in vitro анализ показал, что TNF-α действительно усиливается при активации Т-клеток, где CD4 + и CD8 + Т-лимфоциты стимулировались анти-CD3[8][37] что позже было подтверждено на ранней стадии испытаний с участием солидные опухоли и воспалительные дерматологические заболевания.[38]Ил-12 представляет собой еще один цитокин, который как подавляется, так и усиливается талидомидом и его аналогами. Когда моноциты стимулируются липополисахариды, Продукция ИЛ-12 подавляется, но во время Стимуляция Т-клеток производство расширено.[37]
Считается, что леналидомид примерно в 1000 раз эффективнее in vitro чем талидомид по противовоспалительным свойствам, а помалидомид примерно в 10 раз сильнее, чем леналидомид. Однако стоит отметить, что при сравнении леналидомида и помалидомида клиническая значимость более высокой активности in vitro неясна, поскольку максимально переносимая доза Помалидомид составляет 2 мг в день по сравнению с 25 мг для леналидомида, что приводит к 10-100 раз более низкой концентрации лекарственного средства помалидомида в плазме крови.[39]
Активация Т-клеток
Талидомид и его аналоги помогают костимуляции Т-клеток через B7 -CD28 комплекс путем фосфорилирования тирозин на рецепторе CD28.[8] В пробирке данные свидетельствуют о том, что эта совместная стимуляция приводит к увеличению Чт1 высвобождение цитокинов типа IFN-γ и IL-2, которые дополнительно стимулируют пролиферацию клональных Т-клеток и естественная клетка-убийца распространение и активность. Это усиливает естественные и зависимые от антител клеточные цитотоксичность.[40] Леналидомид и помалидомид примерно в 100-1000 раз более эффективны в стимуляции клональной пролиферации Т-клеток, чем талидомид. Кроме того, in vitro данные показывают, что помалидомид восстанавливает Чт2 клетки в Th1 за счет усиления фактора транскрипции Т-ставка.[32]
Антиангиогенез
Сообщалось, что ангиогенез или рост новых кровеносных сосудов соответствуют прогрессированию ММ, когда фактор роста эндотелия сосудов (VEGF) и его рецептор, bFGF[8] и Ил-6[37] по-видимому, необходимы для миграции эндотелиальных клеток во время ангиогенеза. Считается, что талидомид и его аналоги подавляют ангиогенез за счет модуляции вышеупомянутых факторов, при этом эффективность антиангиогенной активности леналидомида и помалидомида была в 2-3 раза выше, чем у талидомида в различных in vivo анализы,[41] Также было показано, что талидомид блокирует NF-κB активность за счет блокирования IL-6, и было показано, что NF-κB участвует в ангиогенезе.[37] Ингибирование TNF-α не является механизмом ингибирования ангиогенеза талидомидом, поскольку многие другие ингибиторы TNF-α не ингибируют ангиогенез.[6]
Противоопухолевая активность
В естественных условиях Считается, что противоопухолевая активность талидомида обусловлена мощным антиангиогенным действием, а также изменениями экспрессии цитокинов. В пробирке анализы на апоптоз в клетках ММ при обработке талидомидом и его аналогами повышают активность каспаза-8. Это вызывает перекрестный обмен сигналами апоптоза между каспазой-8 и каспаза-9 что приводит к непрямой активации каспазы-9.[32][38] Дальнейшая противоопухолевая активность опосредована ингибированием белка апоптоза-2.[41] и эффекты выживания IGF-1, повышая чувствительность к Опосредованная FAS клеточная смерть и усиление Связанный с TNF лиганд, индуцирующий апоптоз.[38] Также было показано, что они вызывают дозозависимую G0 /G1 клеточный цикл остановка клеточных линий лейкемии[37] где аналоги показали в 100 раз большую эффективность, чем талидомид.[39]
Среда костного мозга
Роль ангиогенеза в поддержании милеомы была впервые обнаружена Ваккой в 1994 году.[42] Они обнаружили, что усиление ангиогенеза костного мозга коррелирует с ростом миеломы, а поддерживающие стромальные клетки являются важным источником ангиогенных молекул при миеломе. Считается, что это главный компонент механизма in vivo с помощью которого талидомид подавляет множественную миелому.
Кроме того, считается, что воспалительные реакции в костном мозге способствуют развитию многих гематологических заболеваний. Секреция ИЛ-6 Костный мозг стромальные клетки (BMSC) и секреция молекул адгезии VCAM-1, ICAM-1 и LFA, индуцируется в присутствии TNF-α и адгезии клеток MM к BMSC. IL-6 способствует пролиферации клеточных линий MM и ингибированию Fas-опосредованного апоптоза.[38] Талидомид и его аналоги непосредственно снижают повышающую регуляцию IL-6 и опосредованно через TNF-α, тем самым снижая секрецию молекул адгезии, что приводит к меньшему количеству клеток MM, прикрепляющихся к BMSC. Остеокласты становятся очень активными во время ММ, что приводит к резорбция кости и секреция различных факторов выживания ММ. Они снижают уровень молекулы адгезии имеет первостепенное значение для активации остеокластов, уменьшает образование клеток, образующих остеокласты, и подавляет катепсин К, важно цистеиновая протеаза экспрессируется в остеокластах.[41]
Апремиласт
In vitro апремиласт снижает PDE4 деятельность, ведущая к увеличению циклический аденозинмонофосфат (цАМФ) в типах иммунных и неиммунных клеток, частично подавляя продукцию многих провоспалительных цитокинов, таких как TNF-α, IFN-γ, IL-2, IL-12 и Ил-23 и повышение продукции противовоспалительного цитокина IL-10.[29][43] Сила подавления продукции TNF-α апремиластом аналогична леналидомиду.[44]
Связь структура-деятельность

Поскольку механизм действия талидомида и его аналогов не совсем ясен, а биорецептор этих веществ не идентифицирован, понимание взаимосвязи между структурой и активностью талидомида и его аналогов в основном основано на молекулярное моделирование и продолжили исследовательское расследование.[17][45]Информация о SAR талидомида и его аналогов все еще обрабатывается, поэтому любые тенденции, подробно описанные здесь, наблюдаются в ходе отдельных исследований. Исследования в основном были сосредоточены на улучшении ингибирования талидомида TNF-α и PDE4.[8][15] а также активность против ангиогенеза.[46][47]
Ингибиторы TNF-α (не через PDE4)
Исследования показали, что замена во фталоильном кольце увеличивает ингибирующую активность TNF-α (рис. 5). Замещение аминогруппы проверяли в различных местах фталоильного кольца (C4, C5, C6, C7) талидомида и ЕМ-12 (описанного ранее). Добавление аминогруппы в месте C4 как на талидомиде, так и на ЕМ-12 привело к гораздо более сильному ингибированию TNF-α. Это также показало, что аминогруппа должна находиться прямо напротив карбонильной группы в изоиндолиноновой кольцевой системе для наиболее сильной активности.[48] Эти аналоги не ингибируют PDE4 и, следовательно, не действуют путем ингибирования PDE4. Другие добавления более длинных и больших групп в положения C4 и C5 фталоильной кольцевой системы талидомида, некоторые с олефин функциональность, были протестированы с различными результатами. Повышенный ингибирующий эффект по сравнению с талидомидом был отмечен для групп, у которых атом кислорода был присоединен непосредственно к олефину C5 или C4. Йод и бром добавление по C4 или C5 привело к равной или пониженной активности по сравнению с талидомидом.[49] Эти группы не сравнивались с леналидомидом или помалидомидом.
Ингибиторы ФДЭ4


Обычная структура аналогов, которые ингибируют TNF-α посредством ингибирования PDE4, получают на основе гидролиза глутаримидного кольца талидомида. Эти аналоги не содержат кислого хирального водорода, в отличие от талидомида, и поэтому можно ожидать, что они будут хирально стабильными.[16]
На фенильном кольце 3,4-диалкоксифенильный фрагмент (фиг.6) является известным фармакофором в ингибиторах PDE4, таких как ролипрам. Оптимальная активность достигается с метоксигруппой в 4-м положении (X2) и большей группой, такой как циклопентокси в 3-м положении углерода (X3). Однако аналоги талидомида, ингибирующие PDE4, не соответствуют SAR аналогов ролипрама. Для аналогов талидомида этоксигруппа в X3 и метоксигруппа в X2, где X1 представляет собой просто водород, давали самое высокое ингибирование PDE4 и TNF-α.[15] Заменители большего размера, чем диэтокси, в положении X2 – X3 имели пониженную активность. Эффекты этих замен, по-видимому, опосредованы стерическими эффектами.[16]
Для позиции Y был исследован ряд групп. Замещенные амиды, которые были крупнее метиламида (CONHCH3) снижают активность ингибирования PDE4.[16] Используя карбоновую кислоту в качестве отправной точки, амидная группа имеет аналогичную активность ингибирования PDE4, но обе группы, как было показано, были значительно менее эффективными, чем группа сложного метилового эфира, у которой было примерно шестикратное увеличение ингибирующей активности PDE4. Сульфоновая группа имела такое же ингибирование PDE4, что и группа метилового эфира. Наилучшее ингибирование PDE4 наблюдалось при присоединении нитрильной группы, которая имеет в 32 раза большую ингибирующую активность PDE4, чем карбоксильная кислота.[15] Заместители в Y, приводящие к увеличению ингибирующей активности PDE4, таким образом, следовали в следующем порядке:
- COOH ≤ CONH2 ≤ КУХНЯ3 ≤ SO2CH3
Были изучены замены во фталоильном кольце, и было замечено, что нитрогруппы в месте C4 или C5 снижали активность, но замещение C4 или C5 амино резко увеличивало ее.[16] Когда было исследовано замещение в положении 4 (Z) на фталоильном кольце, гидроксильные и метоксигруппы, по-видимому, делают аналог менее мощным ингибитором PDE4. Увеличение активности наблюдали с амино и диметиламино в аналогичной степени, но метильная группа улучшала активность больше, чем вышеупомянутые группы. А 4-N-ацетиламиногруппа имела немного более низкую ингибирующую активность PDE4 по сравнению с метильной группой, но увеличивала ингибирующую активность соединения TNF-α в еще большей степени.[15] Заместители в Z, приводящие к увеличению ингибирующей активности PDE4, таким образом, следовали в следующем порядке:
- N (CH3)2 ≤ NH2
3
Подавление ангиогенеза

Для активности ингибирования ангиогенеза неповрежденный глютаримид кольцо вроде требуется. Различные группы были протестированы в позиции R. Вещества, содержащие соли азота в качестве R-группы, показали хорошую активность. Улучшенная ингибирующая активность ангиогенеза может быть обусловлена повышенной растворимостью или тем, что положительно заряженный азот добавил взаимодействие с активным сайтом. Тетрафторирование фталоильного кольца, по-видимому, увеличивает ингибирование ангиогенеза.[46]
Синтез
Эта секция может быть слишком техническим для большинства читателей, чтобы понять. (Апрель 2017 г.) (Узнайте, как и когда удалить этот шаблон сообщения) |
Ниже описаны схемы для синтезирующий талидомид, леналидомид, помалидомид и апремиласт, по данным известных первичная литература. Обратите внимание, что эти схемы синтеза не обязательно отражают стратегии органического синтеза, используемые для синтеза этих отдельных химических соединений.
Талидомид


Синтез талидомида обычно проводят, как показано на схеме 1. Этот синтез представляет собой достаточно упрощенный трехступенчатый процесс. Обратной стороной этого процесса, однако, является то, что последний этап требует высокотемпературной реакции расплава, которая требует нескольких перекристаллизация и не соответствует стандартному оборудованию.
Схема 2 представляет собой новый путь синтеза, который был разработан для того, чтобы сделать реакцию более прямой и обеспечить более высокие выходы. Этот маршрут использует L-глютамин скорее, чем L-глютаминовая кислота в качестве исходного материала и позволяя ему реагировать с N-карбетоксифталимид дает N-фталоил-L-глутамин (4) с выходом 50–70%. Затем вещество 4 перемешивают в смеси с карбонилдиимидазолом (CDI ) с достаточным количеством 4-диметиламинопиридина (DMAP ) в тетрагидрофуране (THF ), чтобы катализировать реакцию и нагревают до рефлюкс в течение 15–18 часов. При кипячении из смеси выкристаллизовывается талидомид. Заключительный этап дает выход талидомида 85–93%, в результате чего общий выход составляет 43–63%.[50]
Леналидомид и помалидомид

Оба аминоаналога получают конденсацией гидрохлорида 3-аминопиперидин-2,6-диона (соединение 3), который синтезируется в двухстадийной реакции из коммерчески доступных КБЗ -L-глютамин. КБЗ-L-глутамин обрабатывают CDI в кипящем с обратным холодильником THF с получением Cbz-аминоглутаримида. Для удаления защитной группы Cbz гидрогенолиз, до 50–60 лет psi водорода с 10% Pd / C смешанный с ацетат этила и HCl. Полученный гидрохлорид (соединение 3 на схеме 3) затем подвергали взаимодействию с 3-нитрофталевым ангидридом в кипящей уксусной кислоте с получением 4-нитрозамещенного аналога талидомида и нитрогруппы, затем восстанавливали с помощью гидрирование дать помалидомид.[48]

Леналидомид синтезируется аналогичным образом с использованием соединения 3 (3-аминопиперидин-2,6-дион), обработанного нитрозамещенным метил-2- (бромметил) бензоатом, и гидрирования нитрогруппы.[48]
Апремиласт

При синтезе апремиласта соединение 3 на схеме 5 получают с выходом 41% из соединения 2, обработанного гексаметилдисилазид лития, в смеси с диметилсульфоном лития и эфиратом трифторида бора. Разделение соединения 3 достигалось обработкой его N-ацетил-L-лейцин с образованием 3S. Последний шаг затем использует конденсация из 3-N-ацетиламинофталевый ангидрид с 3S с получением апремиласта (1S) с выходом 75%.[15]
Фармакокинетика
Талидомид
| Талидомид | ||
|---|---|---|
| ТМаксимум [препарат, средство, медикамент] | 4–6 часов по предметам с ММ[51] |  |
| Связывание с белками | 55–65%[52] | |
| Метаболиты | Гидролизованные метаболиты[52] | |
| Период полураспада [т1/2] | 5,5–7,6 часов[52] | |
Леналидомид
| Леналидомид | ||
|---|---|---|
| ТМаксимум [препарат, средство, медикамент] | 0,6–1,5 часа у здоровых людей[53] 0,5–4 часа по предметам с ММ[54] | 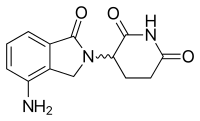 |
| Связывание с белками | ~30%[53] | |
| Метаболиты | Еще не изучено[53] | |
| Период полураспада [т1/2] | 3 часа у здоровых людей[53] 3,1–4,2 часа по предметам с ММ[54] | |
Помалидомид
| Помалидомид | ||
|---|---|---|
| ТМаксимум [препарат, средство, медикамент] | 0,5–8 часов[55] | 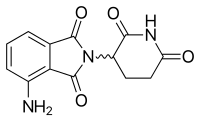 |
| Связывание с белками | Неизвестный | |
| Метаболиты | Неизвестный | |
| Период полураспада [т1/2] | 6,2–7,9 часов[55] | |
Апремиласт
| Апремиласт | ||
|---|---|---|
| ТМаксимум [препарат, средство, медикамент] | 1,5–2 часа у здоровых людей[56] В среднем 2 часа у пациентов с тяжелым псориазом бляшечного типа[57] | 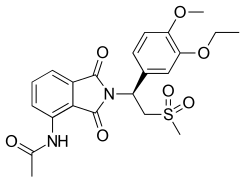 |
| Связывание с белками | ~90%[15] | |
| Метаболиты | О-десметил апремиласт глюкуронид и деметилированный апремиласт вместе с продуктами гидролиза[56] | |
| Период полураспада [т1/2] | 8,2 часов[57] | |
Смотрите также
Рекомендации
- ^ а б c Рыцарь, Р. (август 2005 г.). «IMiDs: новый класс иммуномодуляторов». Семинары по онкологии. 32 (4 Приложение 5): S24 – S30. Дои:10.1053 / j.seminoncol.2005.06.018. PMID 16085014.
- ^ Арагон-Чинг А.Б., Ли Х., Гарднер Э.Р., Фигг В.Д. (2007). «Аналоги талидомида как противоопухолевые препараты». Недавние открытия Pat Anticancer Drug Discov. 2 (2): 167–174. Дои:10.2174/157489207780832478. ЧВК 2048745. PMID 17975653.
- ^ Поворот судьбы: как оскорбленное лекарство стало средством спасения в "войне" против рака - Onco'Zine - Международная сеть онкологов (30 ноября 2013 г.) В архиве 3 января 2014 г., в Archive.today
- ^ Mazzoccoli, L; Кадозо, SH; Amarante, GW; де Соуза, МВ; Домингес, Р. Мачадо, Массачусетс; de Almeida, MV; Тейшейра, ХК (июль 2012 г.). «Новые аналоги талидомида из диаминов подавляют выработку провоспалительных цитокинов и экспрессию CD80, одновременно повышая уровень IL-10». Биомедицина и фармакотерапия. 66 (5): 323–9. Дои:10.1016 / j.biopha.2012.05.001. PMID 22770990.
- ^ Проммер, Э. Э. (20 октября 2009 г.). «Обзорная статья: Паллиативная онкология: талидомид». Американский журнал хосписов и паллиативной медицины. 27 (3): 198–204. Дои:10.1177/1049909109348981. PMID 19843880.
- ^ а б Д'Амато Р.Дж., Локнан М.С., Флинн Э., Фолкман Дж. (Апрель 1994 г.). «Талидомид - ингибитор ангиогенеза». Proc. Natl. Акад. Sci. СОЕДИНЕННЫЕ ШТАТЫ АМЕРИКИ. 91 (9): 4082–5. Дои:10.1073 / пнас.91.9.4082. ЧВК 43727. PMID 7513432.
- ^ Verheul HM, Panigrahy D, Yuan J, D'Amato RJ (январь 1999 г.). «Комбинированная пероральная антиангиогенная терапия с талидомидом и сулиндаком подавляет рост опухоли у кроликов». Br. J. Рак. 79 (1): 114–8. Дои:10.1038 / sj.bjc.6690020. ЧВК 2362163. PMID 10408702.
- ^ а б c d е ж грамм час я j k л Бартлетт, Дж. Блейк; Дредж, Кейт; Далглиш, Ангус Г. (1 апреля 2004 г.). «Хронология: эволюция талидомида и его производных IMiD как противораковых агентов». Обзоры природы Рак. 4 (4): 314–322. Дои:10.1038 / nrc1323. PMID 15057291.
- ^ а б Д'Амато Р.Дж., Ленч С., Андерсон К.С., Роджерс М.С. (декабрь 2001 г.). «Механизм действия талидомида и 3-аминоталидомида при множественной миеломе». Семин. Онкол. 28 (6): 597–601. Дои:10.1016 / S0093-7754 (01) 90031-4. PMID 11740816.
- ^ Циммерман, Тодд (1 мая 2009 г.). «Иммуномодуляторы в онкологии». Обновленная информация о лечении рака. 3 (4): 170–181. Дои:10.1016 / j.uct.2009.03.003.
- ^ Zeldis, Jerome B .; Рыцарь, Роберт; Хусейн, Мохамад; Чопра, Раджеш; Мюллер, Джордж (1 марта 2011 г.). «Обзор истории, свойств и использования иммуномодулирующего соединения леналидомид». Летопись Нью-Йоркской академии наук. 1222 (1): 76–82. Дои:10.1111 / j.1749-6632.2011.05974.x. PMID 21434945.
- ^ http://vectorblog.org/2013/04/from-thalidomide-to-pomalyst-better-living-through-chemistry/
- ^ а б Д'Амато, Р.Дж.; Lentzsch, S; Андерсон, KC; Роджерс, MS (декабрь 2001 г.). «Механизм действия талидомида и 3-аминоталидомида при множественной миеломе». Семинары по онкологии. 28 (6): 597–601. Дои:10.1016 / S0093-7754 (01) 90031-4. PMID 11740816.
- ^ Lentzsch S, Rogers MS, LeBlanc R, et al. (Апрель 2002 г.). «S-3-амино-фталимидо-глутаримид подавляет ангиогенез и рост B-клеточных неоплазий у мышей». Рак Res. 62 (8): 2300–5. PMID 11956087.
- ^ а б c d е ж грамм час я j k Человек, Хон-Ва; Шафер, Питер; Вонг, Лу Минь; Паттерсон, Ребекка Т .; Corral, Laura G .; Раймон, Хизер; Близ, Кейт; Лейстен, Джим; Ширли, Майкл А .; Тан, Ян; Babusis, Darius M .; Чен, Роджер; Стирлинг, Дэйв; Мюллер, Джордж У. (26 марта 2009 г.). "Открытие (S)-N- {2- [1- (3-этокси-4-метоксифенил) -2-метансульфонилэтил] -1,3-диоксо-2,3-дигидро-1ЧАС-изоиндол-4-ил} ацетамид (апремиласт), сильнодействующий и активный при пероральном приеме ингибитор фосфодиэстеразы 4 и фактора некроза опухоли-α ». Журнал медицинской химии. 52 (6): 1522–4. Дои:10.1021 / jm900210d. PMID 19256507.
- ^ а б c d е ж грамм час я j Мюллер, Джордж У .; Corral, Laura G .; Шир, Мэри Дж .; Ван, Хуа; Морейра, Андре; Каплан, Гилла; Стирлинг, Дэвид И. (1 января 1996 г.). «Структурные модификации талидомида производят аналоги с повышенной активностью ингибирования фактора некроза опухоли». Журнал медицинской химии. 39 (17): 3238–3240. Дои:10.1021 / jm9603328. PMID 8765505.
- ^ а б c d е Человек, Хон-Ва; Corral, Laura G; Стирлинг, Дэвид I; Мюллер, Джордж В. (1 октября 2003 г.). «α-Фторзамещенные аналоги талидомида». Письма по биоорганической и медицинской химии. 13 (20): 3415–3417. Дои:10.1016 / S0960-894X (03) 00778-9. PMID 14505639.
- ^ Пан, В; Lentzsch, S (октябрь 2012 г.). «Применение и биология иммуномодулирующих препаратов (IMiD) при раке». Фармакология и терапия. 136 (1): 56–68. Дои:10.1016 / j.pharmthera.2012.07.004. PMID 22796518.
- ^ Седларикова, Л; Кубичкова, Л; Севчикова, С; Хайек, Р. (октябрь 2012 г.). «Механизм действия иммуномодулирующих препаратов при миеломной болезни». Исследование лейкемии. 36 (10): 1218–1224. Дои:10.1016 / j.leukres.2012.05.010. PMID 22727252.
- ^ а б c d Валле, S; Witzens-Harig, M; Jaeger, D; Подар, К (март 2012). «Последняя информация об иммуномодулирующих препаратах (IMiD) при гематологических и солидных злокачественных новообразованиях». Мнение эксперта по фармакотерапии. 13 (4): 473–494. Дои:10.1517/14656566.2012.656091. PMID 22324734.
- ^ «Дозирование таломида (талидомида), показания, взаимодействия, побочные эффекты и многое другое». Ссылка на MedScape. Получено 18 сентября 2012.
- ^ «Талидомид Целген (ранее Талидомид Фармион)». Европейское агентство по лекарствам. Получено 18 сентября 2012.
- ^ «Celgene Biopharmaceutical - Связи с инвесторами - Пресс-релизы». Архивировано из оригинал 19 января 2013 г.. Получено 18 сентября 2012.
- ^ «Дозировка ревлимида (леналидомида), показания, взаимодействия, побочные эффекты и многое другое». Ссылки Medscape. Получено 18 сентября 2012.
- ^ "Поиск: леналидомид - Список результатов". Клинические испытания. Получено 18 сентября 2012.
- ^ «Регистр клинических исследований». Реестр клинических испытаний ЕС. Получено 18 сентября 2012.
- ^ «Celgene отправляет помалидомид на одобрение FDA». Маяк миеломы.
- ^ «Европейское агентство по лекарственным средствам - Результаты поиска по вашему запросу». Европейское агентство по лекарствам. Получено 18 сентября 2012.
- ^ а б Шафер, Питер (1 июня 2012 г.). «Механизм действия и применение апремиласта при псориазе и псориатическом артрите». Биохимическая фармакология. 83 (12): 1583–1590. Дои:10.1016 / j.bcp.2012.01.001. PMID 22257911.
- ^ «FDA одобрило Otezla для лечения псориатического артрита». fda.gov. Получено 13 июн 2014.
- ^ а б Ито, Такуми; Ханда, Хироши (1 марта 2012 г.). «Расшифровка тайны тератогенности талидомида». Врожденные аномалии. 52 (1): 1–7. Дои:10.1111 / j.1741-4520.2011.00351.x. PMID 22348778.
- ^ а б c d е Мартиниани, Роберта; Ди Лорето, Валентина; Ди Сано, Кьяра; Ломбардо, Алессандра; Либерати, Анна Марина (1 января 2012 г.). «Биологическая активность леналидомида и лежащие в его основе терапевтические эффекты при множественной миеломе». Достижения в гематологии. 2012: 842945. Дои:10.1155/2012/842945. ЧВК 3417169. PMID 22919394.
- ^ Quach, H; Ричи, Д; Стюарт, AK; Neeson, P; Харрисон, S; Смит, MJ; Принц, HM (январь 2010 г.). «Механизм действия иммуномодулирующих препаратов (IMiDS) при множественной миеломе». Лейкемия. 24 (1): 22–32. Дои:10.1038 / leu.2009.236. ЧВК 3922408. PMID 19907437.
- ^ Андхаварапу, S; Рой, V (февраль 2013 г.). «Иммуномодулирующие препараты при множественной миеломе». Экспертная оценка гематологии. 6 (1): 69–82. Дои:10.1586 / ehm.12.62. PMID 23373782.
- ^ Седларикова, Л; Кубичкова, Л; Севчикова, С; Хайек, Р. (октябрь 2012 г.). «Механизм действия иммуномодулирующих препаратов при множественной миеломе». Исследование лейкемии. 36 (10): 1218–1224. Дои:10.1016 / j.leukres.2012.05.010. PMID 22727252.
- ^ Чанг, XB; Стюарт, АК (2011). «Какова функциональная роль церблона, связывающего талидомид?». Международный журнал биохимии и молекулярной биологии. 2 (3): 287–94. ЧВК 3193296. PMID 22003441.
- ^ а б c d е ж Хуанг, Йен-Та; Hsu, Chih W .; Чиу, Тед Х. (1 сентября 2008 г.). «Талидомид и его аналоги как противораковые средства». Медицинский журнал Цзы Чи. 20 (3): 188–195. Дои:10.1016 / S1016-3190 (08) 60034-8.
- ^ а б c d е Мельхерт, Магда; Лист, Алан (1 июля 2007 г.). «Сага о талидомиде». Международный журнал биохимии и клеточной биологии. 39 (7–8): 1489–1499. Дои:10.1016 / j.biocel.2007.01.022.
- ^ а б Quach, H; Ричи, Д; Стюарт, A K; Neeson, P; Харрисон, S; Смит, М. Дж .; Принц, HM (12 ноября 2009 г.). «Механизм действия иммуномодулирующих препаратов (IMiDS) при множественной миеломе». Лейкемия. 24 (1): 22–32. Дои:10.1038 / leu.2009.236. ЧВК 3922408. PMID 19907437.
- ^ Thomas, Sheeba K .; Richards, Tiffany A .; Вебер, Донна М. (1 декабря 2007 г.). «Леналидомид при миеломной болезни». Передовая практика и исследования в клинической гематологии. 20 (4): 717–735. Дои:10.1016 / j.beha.2007.09.002. PMID 18070715.
- ^ а б c Котла, Венумадхав; Гоэль, Свати; Нишал, Сангита; Хейк, Кристоф; Вивек, Кумар; Дас, Бхаскар; Верма, Амит (1 января 2009 г.). «Механизм действия леналидомида при гематологических злокачественных новообразованиях». Журнал гематологии и онкологии. 2 (1): 36. Дои:10.1186/1756-8722-2-36. ЧВК 2736171. PMID 19674465.
- ^ Вакка А., Рибатти Д., Ронкали Л. и др. (Июль 1994 г.). «Ангиогенез костного мозга и прогрессирование множественной миеломы». Br. J. Haematol. 87 (3): 503–8. Дои:10.1111 / j.1365-2141.1994.tb08304.x. ЧВК 3301416. PMID 7527645.
- ^ Шафер, PH; Партон, А; Ганди, AK; Капоне, L; Адамс, М; Wu, L; Bartlett, JB; Лавленд, Массачусетс; Гилхар, А; Cheung, Y-F; Бэйли, GS; Houslay, MD; Мужчина, H-W; Мюллер, GW; Стирлинг, Д.И. (1 февраля 2010 г.). «Апремиласт, ингибитор цАМФ фосфодиэстеразы-4, демонстрирует противовоспалительную активность in vitro и на модели псориаза». Британский журнал фармакологии. 159 (4): 842–855. Дои:10.1111 / j.1476-5381.2009.00559.x. ЧВК 2829210. PMID 20050849.
- ^ Мичелли, Миранда Л. (2011). Цирроз печени: причины, диагностика и лечение. Нью-Йорк: Биомедицинские книги Нова. ISBN 978-1-61209-248-5.
- ^ Авила, Каролина Мартинс; Ромейро, Нелильма Коррейя; Sperandio da Silva, Gilberto M .; Sant’Anna, Carlos M.R .; Barreiro, Eliezer J .; Фрага, Карлос А. (1 октября 2006 г.). «Разработка новых моделей CoMFA и CoMSIA 3D-QSAR для противовоспалительных модуляторов TNFα, содержащих фталимид». Биоорганическая и медицинская химия. 14 (20): 6874–6885. Дои:10.1016 / j.bmc.2006.06.042. PMID 16843662.
- ^ а б Леппер, Эрин Р .; Ng, Sylvia S.W .; Гютшов, Майкл; Вайс, Майкл; Хаушильдт, Сунна; Хеккер, Томас К .; Луццио, Фредерик А .; Эгер, Курт; Фигг, Уильям Д. (1 апреля 2004 г.). «Сравнительный анализ молекулярного поля и сравнительный анализ показателей молекулярного сходства аналогов талидомида как ингибиторов ангиогенеза». Журнал медицинской химии. 47 (9): 2219–2227. Дои:10.1021 / jm0304820. PMID 15084120.
- ^ Ногучи, Томоми; Фудзимото, Харука; Сано, Хироко; Миядзима, Ацуши; Миячи, Хироюки; Хашимото, Юичи (1 декабря 2005 г.). «Ингибиторы ангиогенеза, полученные из талидомида». Письма по биоорганической и медицинской химии. 15 (24): 5509–5513. Дои:10.1016 / j.bmcl.2005.08.086. PMID 16183272.
- ^ а б c Мюллер, GW; Chen, R; Huang, SY; Corral, LG; Вонг, Л. М.; Паттерсон, RT; Чен, Y; Каплан, G; Стирлинг, Д.И. (7 июня 1999 г.). «Аминозамещенные аналоги талидомида: мощные ингибиторы производства TNF-альфа». Письма по биоорганической и медицинской химии. 9 (11): 1625–30. Дои:10.1016 / s0960-894x (99) 00250-4. PMID 10386948.
- ^ Стюарт, Скотт Дж .; Спаньоло, Даниэль; Поломска, Марта Э .; Грех, Мелвин; Карими, Махдад; Абрахам, Лоуренс Дж. (1 ноября 2007 г.). «Свойства ингибиторов синтеза и экспрессии TNF новых аналогов талидомида, полученных посредством перекрестного связывания Хека». Письма по биоорганической и медицинской химии. 17 (21): 5819–5824. Дои:10.1016 / j.bmcl.2007.08.042. PMID 17851074.
- ^ Мюллер, Джордж В .; Konnecke, William E .; Смит, Элисон М .; Хетани, Викрам Д. (1 марта 1999 г.). «Краткий двухэтапный синтез талидомида». Исследования и разработки в области органических процессов. 3 (2): 139–140. Дои:10.1021 / op980201b.
- ^ Чанг, Ф. (1 сентября 2004 г.). «Фармакокинетика талидомида и образование метаболитов у мышей, кроликов и пациентов с множественной миеломой». Клинические исследования рака. 10 (17): 5949–5956. Дои:10.1158 / 1078-0432.CCR-04-0421. PMID 15355928.
- ^ а б c «Краткое описание характеристик продукта: талидомид Celgene» (PDF). Европейское агентство по лекарствам. Получено 23 сентября 2012.
- ^ а б c d Armoiry, X .; Aulagner, G .; Факон, Т. (1 июня 2008 г.). «Леналидомид в лечении множественной миеломы: обзор». Журнал клинической фармации и терапии. 33 (3): 219–226. Дои:10.1111 / j.1365-2710.2008.00920.x. PMID 18452408.
- ^ а б Ричардсон, П. Г. (12 июля 2002 г.). «Иммуномодулирующий препарат CC-5013 преодолевает лекарственную устойчивость и хорошо переносится пациентами с рецидивирующей множественной миеломой». Кровь. 100 (9): 3063–3067. Дои:10.1182 / кровь-2002-03-0996. PMID 12384400.
- ^ а б Шей, С.А. (15 августа 2004 г.). «Исследование фазы I иммуномодулирующего аналога талидомида, CC-4047, при рецидивирующей или рефрактерной множественной миеломе». Журнал клинической онкологии. 22 (16): 3269–3276. Дои:10.1200 / JCO.2004.10.052. PMID 15249589.
- ^ а б Hoffmann, M; Кумар, G; Schafer, P; Cedzik, D; Капоне, L; Фонг, KL; Гу, Z; Heller, D; Feng, H; Surapaneni, S; Ласкин, О; Ву, А (декабрь 2011 г.). "Диспозиция, метаболизм и массовый баланс [14C] апремиласт после перорального приема ». Ксенобиотика. 41 (12): 1063–75. Дои:10.3109/00498254.2011.604745. ЧВК 3231940. PMID 21859393.
- ^ а б Готтлиб, AB; Стробер, Б; Крюгер, Дж. Г.; Rohane, P; Zeldis, JB; Ху, СС; Кипнис, К. (май 2008 г.). «Открытое пилотное исследование с участием одной руки у пациентов с тяжелым псориазом бляшечного типа, получавших пероральный противовоспалительный агент апремиласт». Текущие медицинские исследования и мнения. 24 (5): 1529–38. Дои:10.1185 / 030079908X301866. PMID 18419879.