Разработка и открытие лекарств СИОЗС - Development and discovery of SSRI drugs
Селективные ингибиторы обратного захвата серотонина, или же ингибитор обратного захвата серотонина (СИОЗС), являются классом химические соединения которые способствовали значительному прогрессу как антидепрессанты где они произвели революцию в лечении депрессия и другие психические расстройства. СИОЗС терапевтически полезны при лечении паническое расстройство (PD), пост-травматическое стрессовое растройство (ПТСР), социальное тревожное расстройство (также известная как социальная фобия), обсессивно-компульсивное расстройство (ОКР), предменструальное дисфорическое расстройство (PMDD) и анорексия. Существуют также клинические доказательства эффективности СИОЗС в лечении негативных симптомов шизофрения и их способность предотвращать сердечно-сосудистые заболевания.[1]
СИОЗС в первую очередь подавляют переносчик серотонина (SERT) в головном мозге и имеют незначительное влияние на переносчик дофамина (DAT) и переносчик норэпинефрина (СЕТЬ). Запрещение привязки нейротрансмиттер, серотонин (5-HT), к SERT приводит к увеличению концентрации 5-HT в синаптическая щель приводит к усилению связывания 5-HT с постсинаптические рецепторы что приводит к улучшению симптомов депрессии.[2]
Сегодня СИОЗС доминируют на рынке антидепрессантов.[1] и рекомендованы Национальный институт здоровья и клинического совершенства (NICE) в качестве лечения депрессии первой линии, потому что обычно у них меньше побочные эффекты чем другие типы антидепрессантов с такой же эффективностью.[3]
История развития
До открытия препаратов СИОЗС лечение расстройства настроения были относительно ограниченными. Однако сейчас есть десятки антидепрессанты на рынке для лечения депрессии.[4] Ингибиторы моноаминоксидазы (MAOI) и трициклические антидепрессанты (ТЦА) были первыми лекарствами, разработанными для лечения депрессии, появившимися еще в начале 1950-х годов. Из-за нежелательного профиля нежелательных эффектов и высокого потенциала токсичность, из-за их неизбирательного фармакологический эффекты, строгие полки были для приема лекарств, что ограничивало их применение.[4][5] Из-за этого исследователи искали другие альтернативы с аналогичной эффективностью, но с меньшим количеством побочных эффектов, например препараты, которые не вызывали нарушений сердечной проводимости при передозировке или имели тенденцию вызывать припадки,[6] что привело к открытию препаратов СИОЗС. СИОЗС являются наиболее значительным классом антидепрессантов, представленных на рынке в последние годы, и сделали одно из важнейших медицинских открытий последних нескольких десятилетий. СИОЗС были первыми лекарствами, которые, вне всяких сомнений, установили патофизиологический роль 5-HT при аффективных заболеваниях и в широком спектре тревожные расстройства. Точно так же они были первыми, кто подтвердил ингибирование нейромедиатора. повторное поглощение как важный терапевтический принцип.[1][7]

СИОЗС - это первый рационально разработанный класс психотропные препараты. Стратегия, лежащая в основе рациональный дизайн лекарств заключается в разработке нового препарата, способного воздействовать на определенные биологическая мишень или, в данном случае, особый нервный участок действия (насосы поглощения, рецепторы), при этом пытаясь избежать воздействия на другие участки действия. Целью такой разработки является создание фармакологические агенты которые более эффективны, безопасны и лучше переносятся, чем старые лекарства.[8] Первоначальный успех был достигнут, когда медицинские химики начали поиск идеального СИОЗС с химический синтез из цимелидин (Рисунок 1) от антигистаминный препарат препарат, средство, медикамент бромфенирамин,[7] которые демонстрировали селективное ингибирование повторного захвата 5-HT с минимальным ингибированием норэпинефрин (NE) повторное поглощение. Что наиболее важно, у зимелидина не было такого профиля побочных эффектов, как у ТЦА, и поэтому он стал шаблоном для SSRI второго поколения.[5] Цимелидин был первым поступившим в продажу СИОЗС, но в нескольких случаях Синдром Гийена-Барре были связаны с использованием этого препарата, что привело к его изъятию с рынка в 1983 году. Впоследствии было обнаружено и продано несколько нетрициклических СИОЗС. Флуоксетин, который был FDA одобрен в 1987 году и обычно считается первым из имеющихся на рынке СИОЗС, проложивший путь для следующего поколения СИОЗС и считающийся своего рода прототипом.[5] Появление на рынке флуоксетина считается чудодейственным препаратом для лечения депрессии, поскольку у него меньше побочных эффектов, более простые стратегии дозирования и больший запас безопасности при использовании передозировки были потреблены, и поэтому у него была лучшая приверженность по сравнению с более старыми антидепрессантами (ТЦА и ИМАО).[5][9] С тех пор количество препаратов в классе СИОЗС увеличилось, и сейчас их шесть (флуоксетин, пароксетин, циталопрам, эсциталопрам, сертралин и флувоксамин ),[4][8] как показано в Таблица 1.
Таблица 1 Препараты СИОЗС используются для лечения депрессии.
| Флуоксетин | Сертралин | Пароксетин | Флувоксамин | Циталопрам | Эсциталопрам | |
|---|---|---|---|---|---|---|
| Фармацевтические формы | Капсулы, растворимые или диспергируемые таблетки, раствор для приема внутрь | Таблетки, концентрат для перорального применения | Таблетки, суспензия для перорального применения | Таблетки, раствор для приема внутрь, капсулы | Таблетки, раствор для приема внутрь | Таблетки, раствор для приема внутрь |
| Имя бренда | Флуоксетин, Фонтекс, Серомекс, Прозак, Депекс, Серонил, Флутоп, Флуктин | Сертраль, Сертралин, Золофт, Люстрал, Асентра, Треслин | Паксетин, Сероксат, Паксил, Пароксат, Аропакс, Дероксат | Luvox | Оропрам, Циталопрам, Ципрамил, Целекса, Ципрам, Цитокс, Сепрам | Эзопрам, Эсциталопрам, Ципралекс, Лексапро, Сероплекс |
| Дата утверждения FDA | 29 декабря 1987 г.[8] | 30 декабря 1991 г.[8] | 29 декабря 1992 г.[8] | 5 декабря 1994 г. | 17 июля 1998 г.[8] | 14 августа 2002 г.[8] |
Механизм действия
Точный механизм антидепрессивной активности СИОЗС остается несколько неопределенным, но ряд биохимический установлены функции, связанные с лечением СИОЗС.[10] СИОЗС в первую очередь подавляют SERT в головном мозге и незначительно влияют на DAT и NET. СИОЗС также обладают меньшим сродством к α1, α2, H1 и мускариновый рецепторов, которые могут объяснить различия в побочных эффектах между ТЦА и СИОЗС.[5]
Хотя СИОЗС быстро попадают в мозг после приема, и их влияние на повторное поглощение 5-HT можно измерить мгновенно, для их получения требуется около 2–4 недель. терапевтические эффекты.[11] СИОЗС обладают очень высоким селективным сродством к SERT и сразу после введения ингибируют SERT.[12][13] Ингибирование SERT связано с антидепрессивной активностью СИОЗС. 70-80% ингибирование SERT обычно необходимо для того, чтобы вызвать антидепрессивный эффект, и более высокая доза не вызывает более сильного антидепрессивного эффекта для средних пациентов. Однако более высокая доза увеличивает частоту и тяжесть побочных эффектов, связанных с чрезмерным ингибированием обратного захвата 5-HT.[5]

СИОЗС предотвращают связывание 5-HT с SERT[5] который предотвращает всасывание 5-HT обратно в конечную точку пресинапса, где он метаболизируется моноаминоксидаза или хранится в секреторные пузырьки.[12] В результате концентрация 5-HT увеличивается в соматодендритной области 5-HT нейрона, но не так сильно в области терминал аксона площадь (продемонстрировано в фигура 2). Это увеличение концентрации 5-HT вызывает десенсибилизация соматодендритного 5-HT1А ауторецепторы. Когда эти 5-HT1А ауторецепторы были подавленный, они больше не будут ограничивать импульсный поток нейрона 5-HT. Включается импульсный поток, и в результате 5-HT высвобождается на терминале аксона. Однако это увеличение 5-HT происходит не быстро по сравнению с увеличением 5-HT в соматодендретической области 5-HT нейрона. Эта задержка вызвана временем, которое требуется 5-HT для подавления 5-HT.1А ауторецепторы и включают нейроимпульсный поток 5-HT нейрона. Эта задержка может объяснить причину, по которой антидепрессанты не сразу влияют на депрессию. Это также может быть причиной того, что антидепрессивные механизмы могут быть связаны с увеличивающимся потоком нейроимпульсов от 5-HT нейронов, когда концентрация 5-HT увеличивается на конце аксона, прежде чем СИОЗС начинают работать должным образом. Когда СИОЗС (1) ингибируют насос обратного захвата, (2) повышают соматодендритный 5-HT, (3) десенсибилизируют соматодендритный 5-HT1А ауторецепторы, (4) включили импульсный поток и (5) увеличили высвобождение 5-HT с конца аксона, последним шагом могла бы быть десенсибилизация постсинаптических 5-HT рецепторов. Эта десенсибилизация может быть причиной уменьшения побочных эффектов СИОЗС, поскольку толерантность развивается.[13]
Побочные эффекты
Хотя СИОЗС обычно хорошо переносятся и имеют многочисленные преимущества перед другими антидепрессантами, они не лишены побочных эффектов. Побочные эффекты СИОЗС обычно предсказуемы, исходя из их фармакологии, и зависят от дозы. К таким побочным эффектам относятся дисфункция желудочно-кишечного тракта (тошнота, понос, дискомфорт в эпигастрии), влияние на Центральная нервная система (ЦНС) (беспокойство, усталость, тремор ), холинолитик последствия (сухость во рту, помутнение зрения, сонливость, затрудненное мочеиспускание) и сексуальная дисфункция (аноргазмия или же задержка эякуляции ). Иногда симптомы сексуальной дисфункции сохраняются после прекращения приема СИОЗС.[14][15][16]Побочные эффекты СИОЗС обычно легкие и временные и вызывают больше дискомфорта, чем серьезную угрозу с точки зрения системной токсичности. Таким образом, профиль побочного действия СИОЗС может дать определенные терапевтические преимущества в управление депрессией.[17]
Фармакология
СИОЗС хорошо всасываются в желудочно-кишечном тракте и достигают пикового уровня в плазме в течение 1-8 часов.[18][19] Во время абсорбции СИОЗС связываются с белками и широко распределяются по всему телу, включая мозг, в то время как они являются липофильными.[20] Метаболизм и выведение происходит в основном в печени.[19] и большинство СИОЗС продуцируют фармакологически активные метаболиты,[21] как показано в таблица 3 среди других фармакологических свойств СИОЗС.
Таблица 3 Сравнительная фармакология СИОЗС
| Препарат, средство, медикамент | тМаксимум (час) | Биодоступность (%) | VD (Л / кг) | Связывание с белками (%) | т1/2 | Метаболизм | Активные метаболиты[22] | Экскреция |
|---|---|---|---|---|---|---|---|---|
| Флуоксетин | 6–8[23] | 60–80[23] | 20–45[23] | 94.5[23] | Острый прием, 1–3 дня. Хронический прием, 4–6 дней. Норфлуоксетин, острое и хроническое введение, 4–16 дней[23][24] | Обширное прохождение через печень в основном за счет CYP2D6 путем десметилирования. Нелинейный фармакокинетический профиль[9][23][25] | Норфлуоксетин | В основном (60%) моча[25] |
| Сертралин | 4.5–8.4[26] | Абсолютная биодоступность у людей не определена.[27] | 20[27] | 98[26] | 25–26 часов[24][26] | Обширное прохождение через печень в первую очередь за счет CYP2B6[26][28] | Десметил-сертралин (ограниченная активность) | Фекалии и моча в равном количестве[28] |
| Пароксетин | 6–10[29] | 30–60 | 3.1–28[29] | 93–95[29] | 21–24 часов[24][29] | Обширное прохождение через печень в первую очередь за счет CYP2D6. Нелинейный фармакокинетический профиль[9][29] | Нет клинически важных метаболитов | Моча (64%) и фекалии (36%) (с желчью)[30] |
| Флувоксамин | 3–8 | 50 | 25 | 77–80 | 15,6 часов | Печень CYP1A2 и CYP3A4 | Нет клинически важных метаболитов | В основном моча |
| Циталопрам | 2–4[31] | 80[31] | 12[31] | 50[22] | 35 часов[24][31] | Печень CYP3A4 и CYP2C19 в основном через N-деметилирование[31] | Десметил-циталопрам | 12–23% в неизмененном виде в моче и 10% в кале[31] |
| Эсциталопрам | 4–5[32] | 80[32] | 12[32] | 56[22] | 27–32 часов[24][32] | Печень CYP3A4 и CYP2C19 в основном через N-деметилирование[32] | (S) -деметилциталопрам. Не имеет клинического значения | 8–10% (эсциталопрама и (S) -деметилциталопрам (S-DCT)) в моче[32] |
тМаксимум = Время достижения пикового уровня в плазме после перорального приема; VD = Объем распределения; т1/2 = Период полувыведения
Структурные и механические различия между СИОЗС
Признано, что как позиция, так и тип замены на ароматный фрагменты соединений SSRI важны для более высокой специфичности к SERT. Галоген заместители на ароматическом кольце, как обнаружено, в значительной степени ответственны за специфичность СИОЗС к SERT, но все СИОЗС имеют в определенных положениях атомы галогена (Таблица 2). Однако для белка SERT структурная основа его специфичности в отношении SSRI плохо изучена. Исследования показали, что все галогены SSRI связываются с одним и тем же галогенсвязывающим карманом (HBP) в белке SERT, и мутация в этом HBP в SERT резко снижает сродство транспортеров к SSRI.[33]
Как упоминалось ранее, SSRI довольно разнородны в том смысле, что они также связываются с гомологичными NET и DAT, хотя и с гораздо более низким сродством, чем с их основной целью SERT. Селективность SSRI для SERT действительно интересна, когда только одного или двух заместителей различных функциональных групп достаточно для преобразования SSRI в ингибитор обратного захвата норадреналина (NRI) с более высоким сродством к NE.[33] Все антидепрессанты SSRI имеют одинаковый механизм действия и, по крайней мере, в 10 раз более избирательны в отношении ингибирования обратного захвата 5-HT, чем для ингибирования обратного захвата NE. Однако, несмотря на общий механизм действия, СИОЗС различаются по своей эффективности и селективности в ингибировании обратного захвата 5-HT, и многие из них оказывают важное влияние на другие переносчики и рецепторы. СИОЗС структурно разнообразны с явными вариациями в их фармакодинамический и фармакокинетический профилей, что приводит к различиям между ними в их полужизни, клиническая активность, побочные эффекты и лекарственные взаимодействия, что объясняет различия в их эффективности и переносимости среди пациентов.[1][8] Однако все СИОЗС клинически одинаковы, когда дело доходит до их эффективности с течением времени.[5]
Таблица 2 Сравнение химических свойств препаратов СИОЗС
| Препарат, средство, медикамент | ИЮПАК-имя | Классификация | Галоген | Специфика |
|---|---|---|---|---|
Флуоксетин 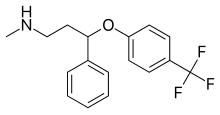 | Метил (3-фенил-3- [4- (трифторметил) фенокси] пропил) амин[23] | Флуоксетин относится к фенилпропиламинам. Они содержат фенилпропиламиновый фрагмент, который состоит из фенильной группы, замещенной у третьего атома углерода пропан-1-амином.[23] | 3F | Наименее селективный ингибитор СИОЗС. Также подавляет обратный захват NE и DA. Также влияет 5-HT2C рецепторы, CYP2D6 и CYP3A4.[8] |
Флувоксамин  | (E) - (2-аминоэтокси) ({5-метокси-1- [4- (трифторметил) фенил] пентилиден}) амин[34] | 3F | ||
Сертралин  | (1S,4S) -4- (3,4-дихлорфенил) -N-метил-1,2,3,4-тетрагидронафталин-1-амин[26] | Сертралин относится к таметралинам. Они содержат фрагмент таметралина, который состоит из тетрагидронафталина, связанного с фенильной группой с образованием N-метил-4-фенил-1,2,3,4-тетрагидронафталин-1-аминный скелет.[26] | 2Cl | Второй по эффективности ингибитор СИОЗС. Также влияет на повторный захват DA и NE.[8] |
Пароксетин  | (3S,4р)-3-[(2ЧАС-1,3-Бензодиоксол-5-илокси) метил] -4- (4-фторфенил) пиперидин[29] | Пароксетин относится к фенилпиперидинам. Они содержат фенилпиперидиновый скелет, который состоит из пиперидина, связанного с фенильной группой.[29] | F | Самый мощный блокатор обратного захвата 5-HT. Это самый мощный блокатор мускариновых рецепторов среди СИОЗС. Также влияет гистамин H1 рецепторы, синтазы оксида азота (БДУ) и CYP2D6.[8] |
Циталопрам  | 1- [3- (диметиламино) пропил] -1- (4-фторфенил) -1,3-дигидро-2-бензофуран-5-карбонитрил[31] | Циталопрам относится к бензофуранам, которые представляют собой органические соединения, содержащие бензольное кольцо, конденсированное с фураном.[31] | F | Второй по селективности ингибитор СИОЗС.[8] |
Эсциталопрам  | (1S) -1- [3- (диметиламино) пропил] -1- (4-фторфенил) -1,3-дигидро-2-бензофуран-5-карбонитрил[32] | Эсциталопрам является S-энантиомером циталопрама и, таким образом, принадлежит к тому же классу бензофуранов, к которому принадлежит циталопрам.[32] | F | Новейший и наиболее селективный ингибитор СИОЗС.[8] |
Взаимосвязь структура-деятельность (SAR)
Производные феноксифенилпропиламина

Соединения, содержащие мотив арилоксипропиламина в своей структуре, продемонстрированной в рисунок 3а, известны как ингибиторы обратного захвата моноаминов. Лекарства, содержащие этот привилегированный структурный мотив, где R1 и R2 находятся арилы или гетероарилы, предпочтительно фенил, обладают профилем селективности в отношении NET и SERT.[35] В то время как соединения, содержащие заместитель в 2'-положении ароксильного кольца структуры (рисунок 3b) проявляет селективность и высокое сродство к NET, и поэтому обычно ИОНИС, соединения, имеющие заместитель в 4'-положении, проявляют селективность и высокое сродство к SERT и поэтому обычно являются SSRI, например флуоксетин и пароксетин.[36]
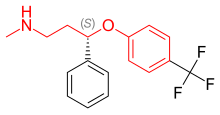
Флуоксетин это рацемическая смесь из (р)- и (S) -флуоксетин, оба энантиомера которого способствуют его биологической активности.[37] Поскольку монозамещение в 4-пара-положении феноксигруппы (фигура 4) приводит к селективному ингибированию повторного захвата 5-HT, дизамещение, то есть 2,3- или 2,4-замена, следовательно, приводит к потере селективности SERT.[5] Флуоксетин имеет самый широкий спектр действия, поскольку он наименее селективен к SERT из всех СИОЗС. Флуоксетин также имеет 5-HT2C антагонистическая активность, когда он блокирует 5-HT активность 5-HT2C рецепторы, усиливающие высвобождение как NE, так и DA. 5-HT2C Антагонисты помогают не только терапевтическими эффектами флуоксетина, но и переносимостью препарата. Преимущество 5-HT2C антагонистом является то, что он оказывает стимулирующее действие, и многие пациенты испытали повышение энергии, концентрации и внимания, а также снижение утомляемости с самой первой дозы. Стимулирующая активность 5-HT2C Однако антагонист может быть недостатком для пациентов с возбуждением, бессонницей и тревогой. Другой особенностью флуоксетина является слабое ингибирование обратного захвата NE, которое может иметь клинический эффект в более высоких дозах. Флуоксетин также имеет длительный период полувыведения, что может уменьшить симптомы отмены, характерные для некоторых СИОЗС после резкого прекращения приема, но это также означает, что для выведения препарата и его активного метаболита после прекращения лечения флуоксетином требуется много времени.[13]

Пароксетин это ограниченный структурный аналог флуоксетина, где линейная фенилпропиламин группа флуоксетина была свернута в пиперидин звенеть (цифра 5). Комплекс имеет возможность четырех стереоизомеры потому что он содержит два хиральные центры, но один из них, (3S,4р) -изомер, продается как пароксетин. Исследования показали, что стереохимические факторы влияют на сродство молекулы к SERT, где замещение в 2-орто-положение любого ароматического кольца снижает сродство к SERT крысы в 10–100 раз, причем наибольшая потеря происходит в феноксильном кольце.[5]
Пароксетин является наиболее сильнодействующим из имеющихся препаратов СИОЗС, но он менее селективен в отношении SERT, чем флувоксамин и сертралин.[38] Пароксетин также имеет слабое ингибирование NET, что может способствовать его эффективности при депрессии, особенно в более высоких дозах. Как показано в Таблица 2Пароксетин также ингибирует фермент NOS, что может быть причиной его неблагоприятного воздействия на сексуальную дисфункцию, особенно у мужчин.[13] Пароксетин показывает самое высокое сродство к мускариновым рецепторам из всех СИОЗС, что приводит к слабой антихолинергической активности и, следовательно, к нежелательным побочным эффектам.[39]

Пока ученые пытались создать новый антидепрессант для ингибирования повторного поглощения NE, они случайно синтезированный два новых соединения, названные талопрам и талсупрам. Эти два соединения не поступали в продажу, несмотря на то, что они являются мощными СИОЗСН, поскольку в ходе клинических испытаний сообщалось о нескольких попытках самоубийства. С небольшими изменениями химической структуры талопрама (рисунок 6), включая единственную замену 6-циано (CN), ученые смогли превратить талопрам в мощный СИОЗС, называемый циталопрам. Но циталопрам также можно рассматривать как ограниченный аналог пароксетина.[5]

Циталопрам занимает второе место по селективности в отношении SERT, не влияет на повторный захват NE или DA и не имеет сродства с другими нейрорецепторами.[5] Циталопрам состоит из двух энантиомеров, (р)- и (S) -, которые являются зеркальным отображением друг друга (рисунок 7). Исследования показали, что почти вся деятельность сосредоточена в (S) -энантиомер и что (р) -циталопрам фактически противодействует действию (S) -энантиомер. Комбинация двух энантиомеров известна как рацемический циталопрам и обладает слабыми антигистаминными свойствами, которые находятся в (р) -энантиомер. Решением для улучшения свойств рацемического циталопрама является удаление нежелательных (р) -энантиомер. Полученный препарат более известен как эсциталопрам, но он состоит только из чистого активного (S) - (+) - изомер. Это изменение, по-видимому, устраняет антигистаминные свойства препарата. Удалив (р) -энантиомер, самая низкая доза эсциталопрама становится более эффективной и начинает действовать быстрее, чем сопоставимая доза циталопрама, где эсциталопрам в два раза активнее циталопрама и по крайней мере в 27 раз сильнее, чем (р) -энантиомер.[5] Таким образом, эсциталопрам является единственным препаратом СИОЗС, для которого чистое ингибирование SERT отвечает почти за все его фармакологические действия. Эсциталопрам - новейший и наиболее селективный ингибитор СИОЗС, и сегодня он считается наиболее переносимым СИОЗС.[5][13]
Производные аминотетралина

Таметралин, соединение, синтезированное в 1978 г. Pfizer Исследования на животных показали, что он является мощным ингибитором обратного захвата NE и DA.[5] Позже было достигнуто неожиданно существенное усиление блокирующей активности захвата 5-HT путем добавления атомов хлора в C-3 и C-4 к структуре таметралина, что привело к (+) -транс-(1р,4S)-N-метил-4-фенил-1-аминотетралин, мощный, но неселективный блокатор захвата. Знак (+) -СНГ-(1S,4S) -изомер, один из четырех диастереомеров соединений, однако проявлял значительно более селективную и сильную ингибирующую активность в отношении захвата 5-HT по сравнению с тремя другими диастереомерами, где 4-фенильное кольцо способствует присоединению в сайтах захвата 5-HT. Соединению присвоено название сертралин (цифра 8).[5][40] Хотя сертралин структурно отличается от других СИОЗС, у него есть фениламинотетралин в его структуре, в которой ядро дифенилпропиламина было вынуждено в жесткое бициклический кольцевая система.[5]
Сертралин является вторым наиболее мощным ингибитором обратного захвата 5-HT, который имеет две очень интересные характеристики, которые его различают, а именно: (1) ингибирующее действие сертралина на DAT и NET и (2) связывание с сигма-1 (σ1) рецептор в ЦНС.[13] Ингибирование DAT и NET является спорным из-за гораздо более слабого ингибирования, которое оно имеет, по сравнению с ингибированием SERT. Сертралин имеет примерно в 60 раз более сильный потенциал ингибирования 5-HT, чем обратный захват NE или DA. Возможно, что требуется лишь умеренное ингибирование DAT и NET, чтобы вызвать повышение энергии, мотивации и концентрации, особенно при добавлении к другой активности, такой как ингибирование SERT.[13] Также было обнаружено, что сертралин обладает высоким сродством к?1 рецепторы. Роль σ1 сайт в фармакологическом действии сертралина может существовать, но значение сродства сертралина для σ1 рецепторы остаются неясными.[41]
Связывание СИОЗС с белком SERT
Молекулярные основы функции СИОЗС, включая их способ связывания и молекулярный механизм ингибирования обратного захвата 5-HT в SERT, полностью не изучены и являются предметом дискуссий. Такая информация очень важна для понимания основных аспектов действия лекарств, начиная от профиля селективности до терапевтической эффективности, и для разработки новых и улучшенных лекарств, нацеленных на SERT человека.[42]
Трехмерная (3D) структура SERT неизвестна и является основным препятствием для выяснения структурного механизма SERT человека. Сравнительная степень молекулярное моделирование были использованы в исследованиях для создания структурных моделей SERT человека в комплексе с его лигандом, но не дали хороших результатов из-за низкого филогенетического и функционального сходства между SERT человека и доступными матричными белками.[42] Однако известна трехмерная структура некоторых бактериальных гомологичных переносчиков, таких как переносчик лейцина (LeuT). Все человеческие SERT, NET и DAT являются членами семейства нейротрансмиттер: симпортер натрия (NSS). SERT содержит примерно 630 аминокислот, которые, по прогнозам, образуют 12 трансмембранные альфа-спирали (TM), которые связаны с внутри- и внеклеточными петлями (IL и EL).[33][43] LeuT, который также является членом семейства NSS, который функционирует как переносчик аминокислот,[33] кристаллизовался из Aquifex aeolicus Ямашита и др.,[44] и разделяет 20-25% идентичности в первичная структура с человеком переносчики нейротрансмиттеров. Следовательно Кристальная структура LeuT и его транспортный механизм оказались хорошей модельной системой для изучения белков NSS.[33] Хотя детальный транспортный механизм белков NSS полностью не изучен, ясно, что для того, чтобы транспорт произошел, должна иметь место перестройка больших белков.[43]
LeuT был совместно кристаллизованный с сертралином и (р)- и (S) -флуоксетин, где было обнаружено, что СИОЗС связываются как неконкурентные ингибиторы в сайте связывания преддверия (может рассматриваться как второй сайт связывания), который отделен от сайта связывания лекарственного средства цепями сайтов двух ароматических аминов. кислоты внеклеточных ворот транспортного белка.[33][43] Все галогены в химической структуре SSRI связываются с одним и тем же HBP в LeuT и взаимодействуют с аналогичными аминокислотами, но аминокислотная последовательность в HBP хорошо сохраняется между LeuT и SERT. Это предполагает, что в человеческом SERT SSRI также связываются как в одном положении, так и аналогичным образом, что является ключевой особенностью, делающей SSRI селективными для SERT. И наоборот, могут быть различия в их связывании, когда другая часть молекулы лекарства, вероятно, будет связываться с SERT по-другому, учитывая разнообразие в их структуре.[33] Локализация сайта вестибулярного связывания, как сайт первичного СИОЗС связывания в SERT является, однако спорной, так как некоторые исследования показали, что работа СИОЗСА в конкурсной основе путем связывания с препаратами связывания сайта, а не на второй сайт связывания.[43]
Связывание флуоксетина с белком LeuT
Оба энантиомера флуоксетина проявляют аналогичное сродство к SERT. Однако селективное соотношение NE: 5HT создает впечатление, что (S) -энантиомер в 100 раз более селективен в отношении ингибирования SERT, чем (р) -энантиомер. (р) - (+) - стереоизомер почти в 8 раз более мощный ингибитор SERT вместе с большей продолжительностью действия, чем (S) - (-) - изомер. (S) - (-) - метаболит норфлуоксетина в семь раз более мощный ингибитор транспортера 5-HT, чем (р) - (+) - метаболит, с коэффициентом селективности, почти эквивалентным (S) -флуоксетин.[5]
Оба энантиомера флуоксетина связываются с внеклеточным вестибулем на белке LeuT таким образом, что три атома фтора метилфеноксильного кольца связываются с HBP, который образуется Leu25, Gly26, Leu29, Arg30 и Tyr108. Галогены дополнительно делают Взаимодействие Ван-дер-Ваальса с Leu29 и Tyr108, где (S) -энантиомер дополнительно связывается с Phe253 и заставляет Ван-дер-Ваальс контактировать с ним среди ранее упомянутых аминокислот. Из-за (S) -энантиомеры, противоположные хиральности (р) -энантиомер, остальная часть молекулы обращена в HBP, где аминный хвост указывает в сторону внеклеточного пространства и взаимодействует с N-конца Leu400, Asp401 и Ala319 (аминокислоты, входящие в состав TM10). В этой форме LeuT-границы комплекс довольно жесткий. Метилфеноксильное кольцо вращается вокруг связи O5-C6 на 46 градусов для (р) -энантиомер и 16 градусов для (S) -энантиомер, но жесткость молекулярной структуры указывает на то, что лекарство сохраняет свою низкоэнергетическую конфигурацию при связывании со своим белком-мишенью.[33]
Связывание сертралина с белком LeuT
Серталин связывается с тем же внеклеточным вестибюлем в LeuT, что и флуоксетин, где два атома хлора на фенильном кольце связываются с HBP, образованным Leu25, Gly26, Leu29, Arg30, Tyr108, Ile111 и Phe253. Галогены дополнительно связывают Ван-дер-Ваальс с Leu29, Tyr108 и Phe253. Тетралин (тетрагидронафталин) на другом конце структуры сертралина находится в контакте с Leu400, Asp401 и Thr409 (которые являются частью TM10), а также молекула взаимодействует с Ala319 петли шпильки EL4 и Arg30 и Gin34 TM1. , где аминный хвост направлен в сторону цитоплазмы. Связанная молекула сертралина имеет дихлорфенильное кольцо, повернутое вокруг связи C4-C13 на 180 градусов по сравнению со свободным лекарственным средством.[33]
Связывание эсциталопрама с белком SERT человека
Андерсен и др. смогли создать модель (S) сайт связывания циталопрама в SERT человека путем комбинирования мутационного анализа и сравнительного моделирования, где они обнаружили, что Asn-177 и Phe-341 являются ключевыми детерминантами для (S) эффективность -циталопрам и ингибирование с высокой аффинностью[42] в дополнение к Tyr-95, Asp-98, Ile-172 и Ser438, описанным ранее, где три функциональные группы в структуре ингибиторов связываются с аминокислотами-переносчиками. (S) -циталопрам позиционируется как цианофталан-. Фторфенильные и метиламиноппильные фрагменты занимают три разных субпакета внутри кармана связывания SERT. Ile-172 и Phe-341, вероятно, не находятся в прямом контакте с молекулой лекарственного средства, но они очень важны для контроля выравнивания ингибитора.[42]
Что было после СИОЗС?
В нейтралитет этой статьи оспаривается. (Июнь 2016) (Узнайте, как и когда удалить этот шаблон сообщения) |
После открытия СИОЗС возрос интерес к новым антидепрессантам с более широким механизмом действия.[нужна цитата ] Венлафаксин (Эффексор) был представлен в 1993 году как первый препарат в ИОНИИ (ингибитор обратного захвата серотонина-норадреналина) класс антидепрессантов. SNRIs отличаются от SSRI тем, что они блокируют повторный захват как 5-HT, так и NE.[45][46] Сегодня СИОЗС наряду с СИОЗС являются наиболее широко используемыми антидепрессантами.[47] В некоторых исследованиях СИОЗС продемонстрировали немного более высокую антидепрессивную эффективность, чем СИОЗС (частота ответа 63,6% против 59,3%).[48] До сих пор ведутся споры о том, являются ли СИОЗС более эффективными, чем СИОЗС.[49]
Смотрите также
- Серотонин
- Селективные ингибиторы обратного захвата серотонина
- Ингибиторы обратного захвата моноаминов
- Антидепрессант второго поколения
- Ингибиторы обратного захвата серотонина-норэпинефрина
Рекомендации
- ^ а б c d Spinks, D .; Спинкс, Г. (2002). Ингибирование обратного захвата серотонина: обновленная информация о текущих исследовательских стратегиях. Современная лекарственная химия. 9. С. 799–810. Дои:10.2174/0929867024606795. ISBN 9781608052042. PMID 11966445. Получено 24 октября 2014.
- ^ Шталь, Стивен М. (1998). «Механизм действия селективных ингибиторов обратного захвата серотонина: рецепторы и пути серотонина опосредуют терапевтические эффекты и побочные эффекты». Журнал аффективных расстройств. 51 (3): 215–235. Дои:10.1016 / S0165-0327 (98) 00221-3. PMID 10333979.
- ^ Национальный институт здоровья и клинического совершенства. «Депрессия у взрослых: лечение и лечение депрессии у взрослых». Национальный институт здоровья и клинического совершенства. Получено 30 октября 2014.
- ^ а б c Фицпатрик, Лаура (07.01.2010). «Краткая история антидепрессантов». Время. Получено 19 октября 2014.
- ^ а б c d е ж грамм час я j k л м п о п q р Lemke, Thomas L .; Уильямс, Дэвид А. (2008). Принципы медицинской химии Фуа (6-е изд.). Филадельфия: Липпинкотт Уильямс и Уилкинс. С. 568–600.
- ^ Фергюсон, Джеймс М. (2001). «Антидепрессанты СИОЗС: побочные эффекты и переносимость». Помощник по первичной медико-санитарной помощи журнала клинической психиатрии. 3 (1): 22–27. Дои:10.4088 / pcc.v03n0105. ЧВК 181155. PMID 15014625.
- ^ а б Карлссон, Арвид. «Открытие СИОЗС: веха в нейропсихофармакологии и рациональном дизайне лекарств» (PDF). landesbioscience.com. Laned Bioscience. Архивировано из оригинал (PDF) на 2014-10-20. Получено 20 октября 2014.
- ^ а б c d е ж грамм час я j k л м «Сравнение селективных ингибиторов обратного захвата серотонина (СИОЗС)». emedexpert.com. eMedExpert. Получено 19 октября 2014.
- ^ а б c Ciraulo, D.A .; Shader, R.I .; Гринблатт, Д.Дж. (2011). Клиническая фармакология и терапия антидепрессантов. Фармакотерапия депрессии, второе издание. С. 33–124. Дои:10.1007/978-1-60327-435-7_2. ISBN 978-1-60327-434-0.
- ^ Фишер Дж. И Ганеллин К. Р. (2010). Открытие лекарств на основе аналогов II. Джон Вили и сыновья. С. 269–270.CS1 maint: несколько имен: список авторов (связь)
- ^ Сарбадхикари, С. Н. (2005). Депрессия и деменция: прогресс в исследованиях мозга, клиническое применение и будущие тенденции. Nova Publishers. п. 195.
- ^ а б Шацберг, А. Ф., и Немерофф, К. Б. (2009). Американский психиатрический учебник психофармакологии.. Американский психиатрический паб. С. 353–355.CS1 maint: несколько имен: список авторов (связь)
- ^ а б c d е ж грамм Шталь, С. М. (2013). Основная психофармакология Шталя: нейробиологические основы и практическое применение. Пресса Кембриджского университета. С. 290–300.
- ^ Бахрик, Одри (2008). «Сохранение побочных эффектов сексуальной дисфункции после прекращения приема антидепрессантов: новые доказательства» (PDF). Журнал открытой психологии. 1: 42–50. Дои:10.2174/1874350100801010042. Архивировано из оригинал (PDF) 19 октября 2013 г.. Получено 30 января 2014.
- ^ Вальдингер, доктор медицины (2015). «Психиатрические расстройства и сексуальная дисфункция». Неврология половых заболеваний и заболеваний мочевого пузыря. Справочник по клинической неврологии / Под редакцией Давида Б. Водушека и Франсуа Боллера. Справочник по клинической неврологии. 130. С. 469–89. Дои:10.1016 / B978-0-444-63247-0.00027-4. ISBN 9780444632470. PMID 26003261.
- ^ http://pi.lilly.com/us/prozac.pdf Стр.14.
- ^ Дэвид Болдуин; Шелдон Прескорн (январь 1995 г.). «СИОЗС: преимущества, недостатки и отличия». Журнал психофармакологии. 9 (2 приложение): 163–178. Дои:10.1177/0269881195009002011. PMID 22297235. S2CID 21474009.
- ^ Zahajszky, J; Rosenbaum, J. F .; Толлефсон, Г. Д. (2009). Американский психиатрический издательский учебник психофармакологии (4-е изд.). Вашингтон, округ Колумбия: American Psychiatric Publishing, Inc., стр. 289.
- ^ а б Прескорн, С. Х. (1997). «Клинически значимая фармакология селективных ингибиторов обратного захвата серотонина - обзор с акцентом на фармакокинетику и влияние на окислительный метаболизм лекарств». Клиническая фармакокинетика.. 32 (Приложение 1): 1–21. Дои:10.2165/00003088-199700321-00003. PMID 9068931. S2CID 43164418.
- ^ Безчлибник-Батлер, Калина З .; Джеффрис, Дж Джоэл (2014). Клинический справочник психотропных препаратов (20-е изд.). Бостон: Издательство Hogrefe. С. 3–14. ISBN 978-1-61676-451-7. Получено 21 октября 2014.
- ^ Aboujaoude, E; Коран, Л. М. (2009). Американский психиатрический учебник по психофармакологии (4-е изд.). Вашингтон, округ Колумбия: American Psychiatric Publishing, Inc., стр. 353.
- ^ а б c Чирауло, Доминик А. (2006). Лекарственные взаимодействия в психиатрии (3-е изд.). Балтимор: Липпинкотт Уильямс и Уилкинс. п. 95. ISBN 9780781748179. Получено 29 октября 2014.
- ^ а б c d е ж грамм час «Флуоксетин». Drugbank.ca. DrugBank. Получено 19 октября 2014.
- ^ а б c d е Хиемке, Кристоф; Харттер, Себастьян (2000). «Фармакокинетика селективных ингибиторов обратного захвата серотонина». Фармакология и терапия. 85 (1): 11–28. Дои:10.1016 / S0163-7258 (99) 00048-0. PMID 10674711.
- ^ а б Европейское агентство по лекарственным средствам. «Краткое описание характеристик продукта» (PDF). Получено 29 октября 2014.
- ^ а б c d е ж «Сертралин». Drugbank.ca. DrugBank. Получено 19 октября 2014.
- ^ а б Сеть токсикологических данных. «Сетралин». Получено 29 октября 2014.
- ^ а б Европейское агентство по лекарственным средствам. «Краткое описание характеристик продукта» (PDF). Получено 29 октября 2014.
- ^ а б c d е ж грамм «Пароксетин». Drugbank.ca. DrugBank. Получено 19 октября 2014.
- ^ Электронный сборник лекарств (eMC). «Краткое описание характеристик продукта». Получено 29 октября 2014.
- ^ а б c d е ж грамм час «Циталопрам». Drugbank.ca. DrugBank. Получено 19 октября 2014.
- ^ а б c d е ж грамм час «Эсциталопрам». Drugbank.ca. DrugBank. Получено 19 октября 2014.
- ^ а б c d е ж грамм час я Чжоу, Чжэн; Чжэнь, Хуан; Карпович, Натан К .; Закон, Кристофер Дж .; Reith, Maarten E.A .; Ван, Да-Ненг (2009). «Антидепрессивная специфичность транспортера серотонина, предложенная тремя структурами LeuT-SSRI». Структурная и молекулярная биология природы. 16 (6): 652–657. Дои:10.1038 / nsmb.1602. ЧВК 2758934. PMID 19430461.
- ^ «Флувоксамин». Drugbank.ca. DrugBank. Получено 19 октября 2014.
- ^ Бут, Джон; и другие. (2005). «Открытие и взаимосвязь между структурой и активностью нового селективного норадреналина и двойных ингибиторов обратного захвата серотонина / норадреналина». Письма по биоорганической и медицинской химии. 15 (3): 699–703. Дои:10.1016 / j.bmcl.2004.11.025. PMID 15664840.
- ^ Mahaney, Paige E .; и другие. (2006). «Синтез и активность нового класса ингибиторов обратного захвата норэпинефрина и серотонина двойного действия: 3- (1H-индол-1-ил) -3-арилпропан-1-аминов». Биоорганическая и медицинская химия. 14 (24): 8455–8466. Дои:10.1016 / j.bmc.2006.08.039. PMID 16973367.
- ^ Оуэнс, Майкл Дж .; Knight, Дэвид Л .; Намерофф, Чарльз Б. (2001). "СИОЗС второго поколения: профиль связывания переносчика моноаминов человека эсциталопрама и R-флуоксетина". Биологическая психиатрия. 50 (5): 345–350. Дои:10.1016 / S0006-3223 (01) 01145-3. PMID 11543737. S2CID 11247427.
- ^ Мозаяни А. и Раймон Л. (2011). Справочник лекарственных взаимодействий: клиническое и судебно-медицинское руководство. Springer. п. 216.CS1 maint: несколько имен: список авторов (связь)
- ^ Fujishiro, J .; Иманиши, Т .; Onozawa, K .; Цусима, М. (2002). «Сравнение антихолинергических эффектов серотонинергических антидепрессантов, пароксетина, флувоксамина и кломипрамина». Европейский журнал фармакологии. 454 (2–3): 183–188. Дои:10.1016 / s0014-2999 (02) 02557-8. PMID 12421645.
- ^ Ко, Б. Кеннет; Вайсман, Альберт; Уэлч, Уиллард М .; Браун, Рональд Г. (1983). «Серталин, 1S, 4S-N-метил-4- (3,4-дихлорфенил) -1,2,3,4-тетрагидро-1-нафтиламин, новый ингибитор поглощения с селективностью в отношении серотонина» (PDF). Журнал фармакологии и экспериментальной терапии. 226 (3): 686–700. PMID 6310078. Архивировано из оригинал (PDF) 4 марта 2016 г.. Получено 22 октября 2014.
- ^ Гленда Маккуин; Лесли Борн; Меир Штайнер (2001). «Селективный ингибитор обратного захвата серотонина сертралин: его профиль и использование при психиатрических расстройствах». Обзоры препаратов для ЦНС. 7 (1): 1–24. Дои:10.1111 / j.1527-3458.2001.tb00188.x. ЧВК 6741657. PMID 11420570.
- ^ а б c d Андерсен, Дж .; Olsen, L .; Hansen, K.B .; Taboureau, O .; Jorgenssen, F.S .; Jorgenssen, A.M .; Bang-Andersen, B .; Egebjerg, J .; Stromgaard, K .; Кристенсен, А. (2010). «Мутационное картирование и моделирование сайта связывания для (S) -циталопрама в транспортере серотонина человека». Журнал биологической химии. 285 (3): 2051–2063. Дои:10.1074 / Jbc.M109.072587. ЧВК 2804362. PMID 19892699.
- ^ а б c d Gabrielsen, M .; Kurczab, R .; Ravna, A.W .; Куфарева, И .; Абагян, Р .; Chilmonczyk, Z .; Bojarski, A.J .; Силте, И. (2012). «Молекулярный механизм ингибирования переносчика серотонина выяснен с помощью нового гибкого протокола стыковки». Европейский журнал медицинской химии. 47 (1): 24–37. Дои:10.1016 / Я.Эймех.2011.09.056. ЧВК 3357065. PMID 22071255.
- ^ Ямасита, А .; Сингх, С.К .; Kawate, T .; Jin, Y .; Гуо, Э. (2005). «Кристаллическая структура бактериального гомолога Na + / Cl - зависимых переносчиков нейромедиаторов». Природа. 437 (7056): 215–223. Дои:10.1038 / природа03978. PMID 16041361. S2CID 4420334.
- ^ Гутьеррес, Массачусетс; Stimmel, GL; Айсо, JY (2003). «Венлафаксин: обновление 2003 года». Клиническая терапия. 25 (8): 2138–54. Дои:10.1016 / s0149-2918 (03) 80210-2. PMID 14512125.
- ^ Гонсалес Руэлас, Энрике; Диас-Мартинес, Алехандро; Мартинес Руис, Рене (1997). «Открытая оценка приемлемости, эффективности и переносимости венлафаксина в обычных условиях оказания медицинской помощи». Текущие терапевтические исследования. 58 (9): 609–630. Дои:10.1016 / S0011-393X (97) 80088-4.
- ^ «200 лучших брендовых препаратов по общему количеству рецептов 2009 г.» (PDF). SDI / Verispan, VONA, полный 2009 г.. www.drugtopics.com. Архивировано из оригинал (PDF) 15 декабря 2012 г.. Получено 6 апреля 2011.
- ^ Папакостас, G .; Thase, M .; Fava, M .; Nelson, J .; Шелтон, Р. (2007). «Являются ли антидепрессанты, сочетающие серотонинергический и норадренергический механизмы действия, более эффективными, чем селективные ингибиторы обратного захвата серотонина, при лечении большого депрессивного расстройства? Метаанализ исследований новых агентов». Биологическая психиатрия. 62 (11): 1217–1227. Дои:10.1016 / j.biopsych.2007.03.027. PMID 17588546. S2CID 45621773.
- ^ Thase, ME (2008). «Являются ли СИОЗС более эффективными, чем СИОЗС? Обзор текущего состояния противоречий». Бюллетень Psychopharmacol. 41 (2): 58–85. PMID 18668017.