Рецептор тромбоксана - Thromboxane receptor




В тромбоксановый рецептор (TP) также известный как простаноидный рецептор TP это белок что у людей кодируется TBXA2R ген, Рецептор тромбоксана является одним из пяти классов простаноидные рецепторы[5] и был первым эйкозаноидный рецептор клонировано.[6] Рецептор TP получил свое название от предпочтительного эндогенного лиганда. тромбоксан А2.[5]
Ген
В ген отвечает за направление синтеза тромбоксанового рецептора, TBXA2R, находится на хромосома 19 в позиции p13.3, пролеты 15 килобазы, и содержит 5 экзоны.[7] TBXA2R коды для члена G-протеин-связанный суперсемейство семи трансмембранных рецепторов.[8][9]
Неоднородность
Молекулярная биология находки предоставили окончательные доказательства для двух подтипов человеческого рецептора TP.[5] Первоначально клонированный ТП из плацента (343 аминокислоты в длину) известен как α изоформа и вариант сращивания клонирован из эндотелий (с 407 аминокислотами), названная β-изоформой.[9] Первые 328 аминокислот одинаковы для обеих изоформ, но β-изоформа имеет расширенный C-концевой цитоплазматический домен.[10] Обе изоформы частично стимулируют клетки за счет активации граммq семейство G-белков.[6] Однако по крайней мере в некоторых типах клеток TPα также стимулирует клетки, активируя G-белки семейства Gs, тогда как TPβ также стимулирует клетки, активируя G-белки класса Gi. Это приводит к стимуляции или ингибированию, соответственно, аденилатциклаза активность и, следовательно, очень разные клеточные реакции.[6] Различия в их С-концевой хвостовой последовательности также допускают значительные различия в интернализации двух рецепторов и, таким образом, десенсибилизации (т. Е. Потери G-белка и, следовательно, способности стимулировать клетки) после активации агонистом; TPβ но не ТПα подвергается интернализации, вызванной агонистами.[11]
Экспрессия изоформ α и β неодинакова в разных типах клеток или между ними.[9] Например, тромбоциты экспрессируют высокие концентрации α-изоформы (и обладают остаточной РНК для β-изоформы), тогда как экспрессия β-изоформы в этих клетках не документирована.[9] Β-изоформа экспрессируется в организме человека. эндотелий.[11] Кроме того, каждая изоформа TP может физически сочетаться с: а) другая его изоформа, чтобы сделать Гомодимеры TPα-TPα или TPβ-TPβ которые способствуют более сильной передаче сигналов в клетках, чем их мономерные аналоги; б) их противоположная изоформа, чтобы сделать Гетеродимеры TPα-TPβ которые активируют больше клеточных сигнальных путей, чем изоформа или гомодимер; и в) с рецептор простациклина (т.е. рецептор IP) с образованием гетеродимеров TP-IP, которые, по сравнению с гетеродимерами TPα-IP, вызывают особенно интенсивную активацию аденилциклаза. Последний эффект на аденилциклазу может служить для подавления стимулирующего действия TPα на клетки и, следовательно, некоторых из его потенциально вредных действий.[12]
Мыши и крысы экспрессируют только изоформу TPα. Поскольку эти грызуны используются в качестве животных моделей для определения функций генов и их продуктов, их отсутствие двух изоформ TP ограничивает понимание индивидуальных и различных функций каждой изоформы рецептора TP.[13]
Распределение тканей
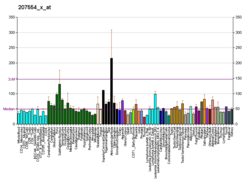
Исторически участию рецептора TP в функции тромбоцитов уделялось наибольшее внимание. Однако теперь ясно, что рецепторы TP обнаруживают широкое распространение в разных типах клеток и среди различных систем органов.[9] Например, рецепторы TP были локализованы, среди прочего, в сердечно-сосудистой, репродуктивной, иммунной, легочной и неврологической тканях.[9][14]
| Орган / Ткань | Клетки / Клеточные линии | |
|---|---|---|
| Распределение рецепторов TP[9] | Легкое, селезенка, матка, плацента, аорта, сердце, кишечник, печень, глаз, тимус, почка, спинной мозг, мозг | Тромбоциты, моноциты крови, мезангиальные клетки клубочков, олигодендроциты, сердечные миоциты, афферентные симпатические нервные окончания в сердце, эпителиальные клетки, клетки Hela, клетки гладкой мускулатуры, эндотелиальные клетки, трофобласты, клетки Шванна, астроциты, мегакариоциты, мегакариоциты, клетки купферролеукта человека, эритроциты человека (HEL), клетки K562 (хронический миелогенный лейкоз человека), клетки гепатобластомы HepG2, незрелые тимоциты, EL-4 (линия Т-клеток мыши), клетки астроцитомы |
Лиганды рецептора TP
Активирующие лиганды
Стандарт простаноиды имеют следующие относительные эффективности как рецепторные лиганды в привязке и активации TP: TXA2=PGH2 >>PGD2 =PGE2 =PGF2alpha =PGI2. Начиная с TXA2 очень нестабилен, связывание рецепторов и биологические исследования TP проводятся со стабильным TXA2 аналоги, такие как I-BOP и U46619. Эти два аналога имеют половину их максимальной связывающей способности и клеточно-стимулирующей активности при ~ 1 и 10-20. наномолярный, соответственно; предполагается, что TXA2 и PGH2 (который также нестабилен) обладают связывающей и клеточно-стимулирующей способностями в этом диапазоне. PGD2, PGE2, PGF2alpha и PGI2 обладают связывающей и стимулирующей способностями, которые в> 1000 раз слабее, чем I-BOP, и поэтому предполагается, что они не обладают заметной способностью стимулировать TP in vivo. 20-гидроксиэйкозатетраеновая кислота (20-HETE) - полный агонист и определенный изопростаны, например 8-изо-PGF2 альфа и 8-изо-PGE2, являются частичные агонисты рецептора TP. В моделях на животных и тканях человека они действуют через TP, чтобы стимулировать реакцию тромбоцитов и стимулировать сокращение кровеносных сосудов.[15] Синтетические аналоги ТХА2 которые активируют TP, но относительно устойчивы к спонтанной и метаболической деградации, включают SQ 26655, AGN192093 и EP 171, каждый из которых обладает связывающей и активирующей способностями к TP, подобными I-BOP.[13][16][17]
Ингибирующие лиганды
Некоторые синтетические соединения связываются с TP, но не активируют, и тем самым ингибируют его активацию, активируя лиганды. Эти антагонисты рецепторов включают I-SAP, SQ-29548, S-145, домитробан и вапипрост, все из которых обладают сродством к связыванию TP, аналогичным I-BOP. Другие известные антагонисты рецепторов TP: Сератродаст (AA-2414), Терутробан (S18886), PTA2, 13-APA, GR-32191, Сулотробан (BM-13177), SQ-29,548, SQ-28,668, ONO-3708, Bay U3405, EP-045, BMS-180,291 и S-145.[5][18] Многие из этих антагонистов рецепторов TP были оценены как потенциальные терапевтические агенты для астма, тромбоз и гипертония.[18] Эти оценки показывают, что антагонисты рецепторов TP могут быть более эффективными, чем лекарства, которые избирательно блокируют выработку TXA.2 ингибиторы тромбоксансинтазы.[18] Этот, казалось бы, парадоксальный результат может отражать способность PGH2, производство которого не блокируется ингибиторами, заменять TXA.2 при активации ТП.[13] Новые антагонисты рецепторов TP, которые также обладают активностью в снижении TXA2 производство путем ингибирования циклооксигеназы были обнаружены и разрабатываются для тестирования на животных моделях.[19]
Механизм клеточной стимуляции
TP классифицируется как сократительный тип простеноидного рецептора на основании его способности сокращать различные типы тканей, содержащих гладкие мышцы, такие как ткани легких, кишечника и матки.[20] TP сокращает гладкую мускулатуру и стимулирует различные реакции в широком диапазоне других клеточных типов путем связывания и мобилизации одного или нескольких семейств G протеин класс рецепторных регулируемых клеточная сигнализация молекулы. При привязке к TXA2, PGH2, или других своих агонистов, TP мобилизует членов:[14][21][22]
- а) Альфа-субъединица Gq семейство (например, типы белков Gq G11, G15 и G16), которое активирует фосфолипаза C, IP3, ячейка Ca2+ мобилизация протеинкиназа Cs, кальмодулин -модулированный киназа легкой цепи миозина, Митоген-активированные протеинкиназы, и Кальциневрин;
- б) G12 / G13 семья, которая активирует Rho GTPases которые контролируют миграцию клеток и движения внутриклеточных органелл;
- в) Альфа-субъединица Gs семья, которая стимулирует аденилциклаза для повышения внутриклеточного уровня лагерь и тем самым активируют цАМФ-регулируемые протеинкиназы A и, таким образом, протеинкиназы A-зависимые клеточная сигнализация пути (см. PKA )
- г) атипичный комплекс белка G Gh / трансглутаминаза-2-кальретикулин что активирует фосфолипаза C, IP3, ячейка Ca2+ мобилизация протеинкиназа C, и Митоген-активированная протеинкиназа но ингибирует аденилциклазу.
После активации этих путей способность рецепторов ТР к клеточной стимуляции быстро меняется на противоположный, что называется процессом. гомологичная десенсибилизация, то есть TP больше не может мобилизовать свои мишени G-белка или дополнительно стимулировать функцию клеток. Впоследствии β, но не α изоформа TP подвергается рецепторная интернализация. Эти события, регулирующие подавление рецепторов вызваны G-протеин-связанные рецепторные киназы мобилизуется во время активации рецептора TP. Агенты, не зависящие от рецептора TP, которые стимулируют клетки к активации протеинкиназы C или же протеинкиназы А может также понижать TP в процессе, называемом гетерологичная десенсибилизация. Например, простациклин I2 (PGI2) -индуцированная активация его рецептор простациклина (IP) и простагландин D2 -индуцированная активация его рецептор простагландина DP1 вызывают десенсибилизацию рецептора TP, активируя протеинкиназы A, в то время как простагландин F2альфа -индуцированная активация его рецептор простагландина F и простагландин E2 -индуцированная активация его рецептор простагландина EP1 рецептор десенсибилизирует TP, активируя протеинкиназы C. Эти десенсибилизирующие ответы служат для ограничения действия агонистов рецептора, а также общей степени возбуждения клеток.[12]
В дополнение к своей способности подавлять TPα рецептор IP активирует сигнальные пути клетки, которые противодействуют тем, которые активируются TP. Кроме того, рецептор IP может физически объединяться с рецептором TPα с образованием гетеродимерного комплекса IP-TPα, который при связывании с TXA2, активирует преимущественно IP-связанные клеточные сигнальные пути. Таким образом, природа и степень многих клеточных ответов на активацию TP-рецептора модулируются IP-рецептором, и эта модуляция может служить для ограничения потенциально вредных эффектов активации TP-рецептора (см. Следующий раздел о функциях).[12][13]
Функции
Исследования с использованием животных, генетически модифицированных с отсутствием рецептора TP, и изучение действия агонистов и антагонистов этого рецептора у животных, а также на ткани животных и человека показывают, что TP выполняет различные функции у животных и что эти функции также выполняются или служат парадигмой для дальнейшего исследование на людях.
Тромбоциты
Человек и животное тромбоциты стимулируется различными агентами, такими как тромбин, продуцирует ТХА2. Ингибирование этого производства значительно снижает конечное количество тромбоцитов. адгезия агрегирование и дегрануляция (то есть секреция содержимого гранул) реагирует на исходный раздражитель. Кроме того, тромбоциты мышей, лишенных рецепторов TP, имеют аналогичные нарушения адгезии, агрегации и дегрануляции, и эти мыши с дефицитом TP не могут образовывать стабильные тромбы и, как следствие, проявляют тенденцию к кровотечению. ТП, как показывают исследования, является частью положительный отзыв петля, которая способствует адгезии тромбоцитов, агрегации, дегрануляции и индуцированной тромбоцитами реакции свертывания крови in vitro и in vivo. Направленные на тромбоциты функции TP во многих отношениях противоположны функциям IP-рецептор. Это дополнительно указывает (см. Предыдущий раздел), что баланс между TXA2-TP и PGI2-IP-оси способствуют регулированию функции тромбоцитов, свертывания крови и кровотечения.[14][13]
Сердечно-сосудистая система
Исследования на животных моделях показывают, что активация рецептора TP сокращает гладкомышечные клетки сосудов и воздействует на сердечные ткани, увеличивая частоту сердечных сокращений, вызывая Сердечные аритмии, и производят миокардиальные ишемия. Эти эффекты могут лежать в основе, по крайней мере частично, защитных эффектов TP. нокаут гена у мышей. TP (- / -) мыши: а) устойчивый к кардиогенный шок вызвано инфузией агониста TP, U46619, или простагландина и тромбоксана A2 предшественник арахидоновая кислота; б) частично защищены от повреждения сердца, вызванного гипертонией в IP -рецепторно-дефицитных мышей кормят высокосолевой диетой; в) предотвращено развитие ангиотензин II -индуцированная гипертензия и гипертензия, индуцированная метиловым эфиром N-нитроаргинина, наряду с ассоциированной гипертрофией сердца; г) устойчивы к повреждению сосудов, вызванному повреждением наружной сонной артерии, вызванным баллонным катетером; д) менее вероятно развитие тяжелой дисфункции микроциркуляции печени, вызванной TNFα а также повреждение почек, вызванное TNFα или бактериальным происхождением эндотоксин; и е) медленно развиваются сосуды атеросклероз в ApoE мыши с нокаутом гена.[12][13][14][23] Кроме того, антагонисты рецепторов TP уменьшают размер инфаркта миокарда в различных моделях этого заболевания на животных и блокируют сердечную дисфункцию, вызванную обширной ишемией тканей на животных моделях дистанционное ишемическое прекондиционирование.[24] Таким образом, TP имеет широкий спектр функций, которые имеют тенденцию быть вредными для сердечно-сосудистой сети у животных и, скорее всего, у людей. Однако функции TP не всегда повреждают сердечно-сосудистую систему: у мышей с истощенными рецепторами TP наблюдается увеличение сердечного повреждения, а также смертность из-за трипаносома круци инфекционное заболевание. Механизм (ы) этого предполагаемого защитного эффекта и его применимость к людям пока не известны.[14]
20-гидроксиэйкозатетраеновая кислота (20-HETE), продукт арахидоновая кислота образована Омега-гидроксилазы цитохрома P450,[25] и некоторые изопростаны, образующиеся неферментативными свободный радикал атака на арахидоновую кислоту,[17] сужать препараты артерий грызунов и человека, непосредственно активируя TP. Хотя он значительно менее эффективен, чем тромбоксан А2 в активации этого рецептора, исследования на препаратах мозговых артерий крыс и человека показывают, что усиление кровотока через эти артерии вызывает выработку 20-НЕТЕ, который, в свою очередь, связывает рецепторы ТР, сужая эти сосуды и тем самым уменьшая их кровообращение. . Предполагается, что выступающий в качестве последнего, 20-HETE функционирует как TXA.2 аналог для регулирования кровотока в головном мозге и, возможно, в других органах.[15][26] Изопростаны образуются в тканях, находящихся в остром или хроническом состоянии. окислительный стресс например, возникает в очагах воспаления и артериях больных диабетом.[17] Высокие уровни изопростанов образуются в ишемизированных или иным образом поврежденных кровеносных сосудах и, действуя через TP, могут стимулировать артериальное воспаление и разрастание гладких мышц; Предполагается, что эта ось изопростан-TP способствует развитию атеросклероза и, следовательно, сердечных приступов и инсультов у людей.[17][19]
Легочная аллергическая реактивность
Активация рецептора TP сокращает препараты гладких мышц бронхов, полученные как на животных моделях, так и на людях, и сокращает дыхательные пути на животных моделях.[14] В модели астмы у мышей (т.е. гиперчувствительности к овалабумину) антагонист рецептора TP уменьшал количество эозинофилов, инфильтрирующих легкие, о чем судили по их содержанию в Бронхоальвеолярный лаваж жидкости и в мышиной модели астмы, вызванной пылевым клещом, удаление TBXA2R предотвращал развитие сужения дыхательных путей и реакции легочной эозинофилии на аллерген. Другие агонисты рецептора TP также снижали реактивность бронхов дыхательных путей к аллергену, а также уменьшали симптомы у добровольцев с астмой.[27] Рецептор TP, по-видимому, играет важную роль в проастматическом действии лейкотриен C4 (LTC4): у мышей, сенсибилизированных овальбумином, лейкотриен C4 увеличивало количество эозинофилов в жидкости бронхоальвеолярного лаважа и одновременно снижало процентное содержание эозинофилов в крови, но эти реакции не возникали в TBXA2R-дефицитные мыши. LTC4 также стимулировал экспрессию в легких провоспалительных молекул внутриклеточной адгезии, ICAM-1 и VCAM-1 по механизму, зависимому от рецептора TP.[28] Эти данные свидетельствуют о том, что TP способствует развитию астмы на животных моделях, по крайней мере, частично, опосредуя действия LTC4. Необходимы дальнейшие исследования, чтобы определить, могут ли антагонисты рецепторов TP быть полезными для лечения астмы и других синдромов сужения дыхательных путей, таких как хронические обструктивные заболевания легких в людях.
Матка
Вместе с PGF2α действуя через Рецептор FP, TXA2 действуя через TP, сокращает препараты гладкой мускулатуры матки грызунов и людей. Поскольку матка человека теряет чувствительность к PGP2α, но не к TXA2 вовремя ранние сроки родов при вагинальных родах Предполагается, что агонисты TP могут быть полезны для лечения преждевременных родов.[14]
Иммунная система
Активация рецепторов TP стимулирует провоспалительные реакции эндотелиальных клеток сосудов, такие как повышенная экспрессия белков адгезии на поверхности клетки (т.е. ICAM-1, VCAM-1, и E-selectin ); стимулирует апоптоз (т.е. смерть клетки) CD4 + и CD8 + лимфоциты; вызывает хемокинез (т.е. движение клеток) родного Т-клетки; и ухудшает адгезию дендритные клетки к Т-клетки тем самым ингибируя зависимую от дендритных клеток пролиферацию Т-клеток. Мыши с дефицитом TP демонстрируют повышенную реакцию контактной гиперчувствительности на DNFB тимоциты в вилочковая железа из этих мышей с дефицитом устойчивы к липополисахарид -индуцированный апоптоз. Мыши с истощенными рецепторами TP также постепенно развиваются с возрастом. лимфаденопатия и, как следствие, усиление иммунного ответа на чужеродные антигены. Эти исследования показывают, что передача сигналов TXA2-TP действует как негативный регулятор взаимодействий DC-T-клеток и, возможно, тем самым приобретение приобретенный иммунитет у мышей. Необходимы дальнейшие исследования, чтобы перенести эти исследования на мышах на людей.[14][29][30]
Рак
Повышенное выражение циклооксигеназы и их потенциальное участие в прогрессировании различных раковых заболеваний человека. Некоторые исследования показывают, что TXA2 последующий метаболит этих циклооксигеназ вместе с его рецептором TP вносят вклад в опосредование этого прогрессирования. Активация TP стимулирует пролиферацию, миграцию опухолевых клеток, неоваскуляризация, инвазивность и метастазирование в моделях животных, моделях клеток животных и человека и / или образцах тканей человека при раке простаты, груди, легких, толстой кишки, мозга и мочевого пузыря.[14][31] Эти результаты, хотя и наводящие на размышления, нуждаются в трансляционных исследованиях, чтобы определить их отношение к указанным видам рака человека.
Клиническое значение
Было обнаружено, что в отдельных случаях у людей со склонностью к кровотечениям от легкой до умеренной степени мутации в TP связаны с дефектами связывания TXA с рецепторами.2 аналоги, активирующие сигнальные пути клеток и / или функциональные ответы тромбоцитов не только на агонисты TP, но также и на агенты, которые стимулируют тромбоциты с помощью TP-независимых механизмов (см. раздел «Геномика» ниже).[15]
Используемые препараты, направленные на ТП
Антагонист рецептора TP Сератродаст продается в Японии и Китае для лечения астмы. Пикотамид, двойной ингибитор TP и TXA2 синтез, лицензирован в Италии для лечения клинических тромбозов артерий и заболеваний периферических артерий.[15] Эти препараты еще не лицензированы для использования в других странах.
Клинические испытания
В то время как функциональная роль передачи сигналов рецептора TP в различных гомеостатических и патологических процессах была продемонстрирована на животных моделях, на людях эта роль была продемонстрирована в основном в отношении функции тромбоцитов, свертывания крови и гемостаз. Также предполагается, что TP участвует в регуляции кровяного давления и кровотока в органах человека; существенный и вызванный беременностью гипертония; сосудистые осложнения из-за серповидноклеточной анемии; другие сердечно-сосудистые заболевания, включая острое сердечно-сосудистое заболевание, Инсульт, и заболевания периферических артерий; сокращение матки при родах; и модуляция врожденных и адаптивных иммунные ответы в том числе вызывающие различные аллергические и воспалительные заболевания кишечника, легких и почек.[9] Тем не менее, многие исследования на животных моделях и тканях, подтверждающие эти предполагаемые функции, еще предстоит доказать, что они напрямую применимы к заболеваниям человека. Исследования, чтобы предоставить эти доказательства, в первую очередь основываются на определении того, являются ли антагонисты рецепторов TP клинически полезными. Однако эти исследования сталкиваются с проблемами, связанными с тем, что препараты, косвенно воздействующие на TP (например, Нестероидные противовоспалительные препараты этот блок TXA2 production) или в обход TP (например, P2Y12 антагонисты, ингибирующие активацию тромбоцитов и кортикостероиды и цистеиниловый лейкотриеновый рецептор 1 антагонисты, подавляющие аллергические и / или воспалительные реакции) являются эффективными средствами лечения многих предположительно TP-зависимых заболеваний. Эти препараты, вероятно, будут дешевле и могут иметь более серьезные побочные эффекты, чем препараты, нацеленные на TP.[14] Эти соображения могут помочь объяснить, почему относительно небольшое количество исследований изучали клиническую ценность лекарств, нацеленных на TP. Следующее переводческие исследования по антагонистам TP были проведены или в процессе:[27][19]
- В нерандомизированном неконтролируемом обследовании 4 недели лечения антагонистом рецепторов TP AA-2414 значительно снизили реактивность бронхов у пациентов с астмой. Продолжение двойное слепое плацебо-контролируемое исследование пациентов с астмой обнаружили, что антагонист рецепторов TP Сератродаст значительно уменьшено поток в дыхательных путях (т.е. ОФВ1), суточные колебания ОФВ1, чувствительность дыхательных путей к сократительной стимуляции, воспаление дыхательных путей и содержание проаллергических медиаторов в дыхательных путях (т.е. RANTES, CCL3, CCL7, и эотаксин ).
- А фаза 3 исследование, антагонист TP Терутробан был протестирован против аспирина как профилактика повторных, а также новых ишемия события у пациентов с недавним удары или же преходящие ишемические атаки. Исследование не достигло своих основных конечных точек по сравнению с контрольной группой, получавшей аспирин, и было остановлено; у пациентов, принимавших препарат, наблюдалось значительное увеличение эпизодов незначительных кровотечений.
- Исследование, сравнивающее безопасность и эффективность антагониста TP ридогрела и аспирина в качестве дополнительной терапии при неотложном лечении сердечного приступа с помощью агента, растворяющего сгустки. стрептокиназа обнаружили, что ридогрель не дает значительного улучшения рассасывания сгустка, но снижает частоту повторных сердечных приступов, рецидивирующих стенокардия, и новые инсульты, не вызывая чрезмерных кровотечений ** осложнений.
- Антагонист TP Ифетробан в фаза 2 клинические разработки для лечения почечной недостаточности.
В дополнение к вышеуказанным антагонистам TP, лекарства, которые обладают двойным ингибирующим действием, поскольку они блокируют не только TP, но также блокируют фермент, ответственный за выработку TXA2.2, Тромбоксан-синтаза, находятся в клинической разработке. Эти исследования двойных ингибиторов включают:[15]
- В долгосрочном исследовании у пациентов с сахарным диабетом сравнивали двойной ингибитор пикотамид аспирину для улучшения симптомов ишемии, вызванной заболевания периферических артерий не обнаружили разницы в первичных конечных точках, но также обнаружили, что терапия пикотамидом значительно снижает сердечно-сосудистую смертность в течение 2-летнего испытания.
- Фаза 2 клинических испытаний двойного ингибитора Тербогрель лечение вазоконстрикции было прекращено из-за индукции боли в ногах.
- Двойной ингибитор EV-077 находится в клинической фазе II разработки.
Геномика
Было обнаружено, что несколько изолированных и / или унаследованных случаев пациентов, страдающих кровоточащим диатезом от легкой до умеренно тяжелой степени, связаны с мутациями в TBXA2R. ген, который приводит к аномалиям в экспрессии, субклеточном расположении или функции его продукта TP. Эти случаи включают:[15][32]
- А миссенс-мутация вызывая замену триптофана (Trp) на цистеин (Cys), поскольку его 29-я аминокислота (например, Trp29Cys) дает TP, который менее чувствителен к стимуляции агонистом TP, менее способен активировать свой белок-мишень Gq G и плохо экспрессируется на поверхность клетки. Некоторые или, возможно, все эти дефекты могут отражать неспособность этого мутировавшего TP образовывать димеры TP-TP.
- Мутация Asn42Ser дает TP, который остается в клетке. аппарат Гольджи и не экспрессируется на поверхности клетки.
- Мутация Asp304Asn дает TP, который демонстрирует пониженное связывание и чувствительность к агонисту TP.
- Мутация Arg60Leu дает TP, который обычно экспрессируется и обычно связывает агонист TP, но не активирует его белок-мишень GqG.
- Миссенс-мутация, при которой тимин (T) заменяется гуанином (G) в качестве 175 нуклеотида (c.175C> T) в TBXA2R ген, а также мутации Cc87G> C и c.125A> G дают ТП, которые плохо известны.
- Мутация c.190G> A дает TP, который плохо связывает агонист TP.
- Дупликация гуанина (G) на 167-м нуклеотиде вызывает Мутация сдвига рамки (c.165dupG) по аминокислоте № 58 с получением плохо экспрессируемого мутанта TP.
Однонуклеотидный полиморфизм (SNP) вариации TBXA2R ген были связаны с аллергическими и сердечно-сосудистыми заболеваниями; к ним относятся:[33][34]
- Мета-анализ нескольких исследований, проведенных на разных популяционных тестовых группах, подтвердили связь TBXA2R однонуклеотидный полиморфизм (SNP) вариант 924C> T с повышенным риском развития астмы. Частота варианта SNP 795T> C в TBXA2R была обнаружена в отдельных исследованиях южнокорейской и японской тестовых групп, а частота варианта SNP -6484C> T, предшествующего TBXA2R ген в исследовании южнокорейской тестовой группы был обнаружен повышенным у пациентов, страдающих тяжелой формой астмы, называемой Аспирин-индуцированная астма. Оба варианта SNP 795T> C и 924C> T кодируют рецептор TP, который проявляет повышенное связывание и чувствительность к TXA.2 аналоги. Вариант SNP -4684T ассоциировался с пониженным активность промотора гена в TBXA2R ген и повышенная частота развития индуцированного аспирином крапивница в корейской тестовой группе.
- Вариант SNP rs768963 в TBX2R был связан с увеличением частоты крупных артерий атеросклероз, окклюзия малой артерии и Инсульт в двух отдельных исследованиях китайских тестовых групп. В одной из последних групп гаплотип T-T-G-T C795T-T924C-G1686A-rs768963 был значительно реже у пациентов, перенесших инсульт. Вариант SNP rs13306046 показал снижение микроРНК -индуцированное подавление TBXA2R экспрессия гена и была связана со снижением артериального давления в скандинавской кавказской тестовой группе.
Смотрите также
Рекомендации
- ^ а б c ГРЧ38: Ансамбль выпуск 89: ENSG00000006638 - Ансамбль, Май 2017
- ^ а б c GRCm38: выпуск Ensembl 89: ENSMUSG00000034881 - Ансамбль, Май 2017
- ^ "Справочник человека по PubMed:". Национальный центр биотехнологической информации, Национальная медицинская библиотека США.
- ^ "Ссылка на Mouse PubMed:". Национальный центр биотехнологической информации, Национальная медицинская библиотека США.
- ^ а б c d Девиллер П., Бессар Г. (1997). "Тромбоксан А2 и родственные простагландины в дыхательных путях ". Fundam Clin Pharmacol. 11 (1): 2–18. Дои:10.1111 / j.1472-8206.1997.tb00163.x. PMID 9182072. S2CID 20514470.
- ^ а б c Rolin S, Masereel B, Dogné JM (март 2006 г.). «Простаноиды как фармакологические мишени при ХОБЛ и астме». Eur J Pharmacol. 533 (1–3): 89–100. Дои:10.1016 / j.ejphar.2005.12.058. PMID 16458293.
- ^ TBXA2R тромбоксановый рецептор A2 (Homo sapiens)
- ^ Абэ Т., Такеучи К., Такахаши Н., Цуцуми Э., Танияма Ю., Абэ К. (1995). «Рецептор тромбоксана почки крысы: молекулярное клонирование, сигнальная трансдукция и локализация внутрипочечной экспрессии». J. Clin. Вкладывать деньги. 96 (2): 657–64. Дои:10.1172 / JCI118108. ЧВК 185246. PMID 7635958.
- ^ а б c d е ж грамм час Хуанг Дж. С., Рамамурти СК, Лин Х, Ле Бретон, GC (май 2004 г.). «Передача сигналов через тромбоксановые рецепторы А2». Сотовый сигнал. 16 (5): 521–33. Дои:10.1016 / j.cellsig.2003.10.008. PMID 14751539.
- ^ Foulon I, Bachir D, Galacteros F, Maclouf J (1993). «Повышенная продукция тромбоксана in vivo у пациентов с серповидно-клеточной анемией сопровождается нарушением функций тромбоцитов агониста тромбоксана A2 U46619». Артериосклероз и тромбоз. 13 (3): 421–6. Дои:10.1161 / 01.atv.13.3.421. PMID 8443146.
- ^ а б Фарук SP, Arm JP, Ли TH (2008). «Липидные медиаторы: лейкотриены, простаноиды, липоксины и фактор, активирующий тромбоциты». В Holt PG, Kaplan AP, Bousquet J (ред.). Аллергия и аллергические заболевания. 1 (2-е изд.). Оксфорд, Великобритания: Wiley-Blackwell. ISBN 978-1-4051-5720-9.
- ^ а б c d Korbecki J, Baranowska-Bosiacka I, Gutowska I, Chlubek D (2014). «Циклооксигеназные пути». Acta Biochimica Polonica. 61 (4): 639–49. Дои:10.18388 / abp.2014_1825. PMID 25343148.
- ^ а б c d е ж Риччиотти Э., Фитцджеральд Г.А. (2011). «Простагландины и воспаление». Артериосклероз, тромбоз и биология сосудов. 31 (5): 986–1000. Дои:10.1161 / ATVBAHA.110.207449. ЧВК 3081099. PMID 21508345.
- ^ а б c d е ж грамм час я j Вудворд Д.Ф., Джонс Р.Л., Нарумия С. (2011). «Международный союз фундаментальной и клинической фармакологии. LXXXIII: классификация простаноидных рецепторов, обновление за 15 лет». Фармакологические обзоры. 63 (3): 471–538. Дои:10.1124 / пр.110.003517. PMID 21752876.
- ^ а б c d е ж Capra V, Bäck M, Angiolillo DJ, Cattaneo M, Sakariassen KS (2014). «Влияние активации простаноидных рецепторов тромбоксана сосудов на гемостаз, тромбоз, окислительный стресс и воспаление». Журнал тромбоза и гемостаза. 12 (2): 126–37. Дои:10.1111 / jth.12472. PMID 24298905. S2CID 26569858.
- ^ http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=346
- ^ а б c d Bauer J, Ripperger A, Frantz S, Ergün S, Schwedhelm E, Benndorf RA (2014). «Патофизиология изопростанов в сердечно-сосудистой системе: последствия активации изопростан-опосредованного рецептора тромбоксана А2». Британский журнал фармакологии. 171 (13): 3115–31. Дои:10.1111 / bph.12677. ЧВК 4080968. PMID 24646155.
- ^ а б c Шен РФ, Тай ХХ (1998). «Тромбоксаны: синтаза и рецепторы». J Biomed Sci. 5 (3): 153–72. Дои:10.1007 / BF02253465. PMID 9678486.
- ^ а б c Ходжа М., Буччеллати С., Капра В., Гарелла Д., Сина С., Роландо Б., Фруттеро Р., Карневали С., Сала А., Ровати Г.Э., Бертинария М. (2016). «Фармакологическая оценка in vitro многоцелевых агентов для антагонизма тромбоксановых простаноидных рецепторов и ингибирования ЦОГ-2» (PDF). Фармакологические исследования. 103: 132–43. Дои:10.1016 / j.phrs.2015.11.012. HDL:2318/1551575. PMID 26621246.
- ^ Мацуока Т., Нарумия С. (2008). «Роль простаноидов в поведении при инфекциях и болезнях». Журнал инфекций и химиотерапии. 14 (4): 270–8. Дои:10.1007 / s10156-008-0622-3. PMID 18709530. S2CID 207058745.
- ^ Mhaouty-Kodja S (2004). «Гальфа / тканевая трансглутаминаза 2: появляющийся G-белок в сигнальной трансдукции». Биология клетки. 96 (5): 363–7. Дои:10.1016 / j.biolcel.2004.03.003. PMID 15207905.
- ^ Пак МК, Чой Дж. К., Ким Х. Дж., Накахата Н., Лим К. М., Ким СИ, Ли Ч. (2014). «Новые ингибирующие эффекты кардамонина на реакцию царапания, вызванную тромбоксаном A2: блокирование связывания Gh / трансглутаминазы-2 с рецептором тромбоксана A2». Фармакология, биохимия и поведение. 126: 131–5. Дои:10.1016 / j.pbb.2014.09.011. PMID 25285619. S2CID 144250159.
- ^ Сильва Б.Р., Паула Т.Д., Пауло М., Бендхак Л.М. (2016). «Передача сигналов оксида азота и перекрестная связь с простаноидными путями в сосудистой системе». Медицинская химия. PMID 28031017.
- ^ Аггарвал С., Рандхава П.К., Сингх Н., Джагги А.С. (2016). «Прекондиционирование на расстоянии: участие эндотелиальных вазоактивных веществ в кардиопротекции против ишемического реперфузионного повреждения». Науки о жизни. 151: 250–8. Дои:10.1016 / j.lfs.2016.03.021. PMID 26979771.
- ^ Кроец Д.Л., Сюй Ф. (2005). «Регулирование и ингибирование омега-гидроксилаз арахидоновой кислоты и образования 20-HETE». Ежегодный обзор фармакологии и токсикологии. 45: 413–38. Дои:10.1146 / annurev.pharmtox.45.120403.100045. PMID 15822183.
- ^ Тот П., Роза Б., Спринго З, Докзи Т., Коллер А. (2011). «Изолированные церебральные артерии человека и крысы сжимаются из-за увеличения кровотока: роль рецепторов 20-HETE и TP». Журнал церебрального кровотока и метаболизма. 31 (10): 2096–105. Дои:10.1038 / jcbfm.2011.74. ЧВК 3208155. PMID 21610722.
- ^ а б Клаар Д., Hartert TV, Peebles RS (2015). «Роль простагландинов при аллергическом воспалении легких и астме». Экспертный обзор респираторной медицины. 9 (1): 55–72. Дои:10.1586/17476348.2015.992783. ЧВК 4380345. PMID 25541289.
- ^ Лю Т., Гарофало Д., Фэн С., Лай Дж., Кац Х., Лейдлоу TM, Бойс Дж. А. (2015). «Воспаление дыхательных путей, вызванное лейкотриеном C4, вызванное тромбоцитами, у мышей является чувствительным к аспирину и зависит от T-простаноидных рецепторов». Журнал иммунологии. 194 (11): 5061–8. Дои:10.4049 / jimmunol.1402959. ЧВК 4433852. PMID 25904552.
- ^ Накахата Н (2008). «Тромбоксан А2: физиология / патофизиология, передача клеточного сигнала и фармакология». Фармакология и терапия. 118 (1): 18–35. Дои:10.1016 / j.pharmthera.2008.01.001. PMID 18374420.
- ^ Саката Д., Яо С., Нарумия С. (2010). «Новые роли простаноидов в Т-клеточном иммунитете». IUBMB Life. 62 (8): 591–6. Дои:10.1002 / iub.356. PMID 20665621. S2CID 9889648.
- ^ Экамбарам П., Ламбив В., Каззолли Р., Эштон А.В., Хонн К.В. (2011). «Тромбоксансинтаза и сигнальный путь рецептора при раке: новая парадигма в развитии рака и метастазировании». Отзывы о метастазах рака. 30 (3–4): 397–408. Дои:10.1007 / s10555-011-9297-9. ЧВК 4175445. PMID 22037941.
- ^ Нисар С.П., Джонс М.Л., Каннингем М.Р., Мамфорд А.Д., Манделл С.Дж. (2015). «Редкие варианты GPCR тромбоцитов: что мы можем узнать?». Британский журнал фармакологии. 172 (13): 3242–53. Дои:10.1111 / bph.12941. ЧВК 4500363. PMID 25231155.
- ^ Корнехо-Гарсия Х.А., Перкинс-младший, Хурадо-Эскобар Р., Гарсия-Мартин Э., Агундес Х.А., Вигера Э., Перес-Санчес Н., Бланка-Лопес Н. (2016). «Фармакогеномика простагландиновых и лейкотриеновых рецепторов». Границы фармакологии. 7: 316. Дои:10.3389 / fphar.2016.00316. ЧВК 5030812. PMID 27708579.
- ^ Томпсон MD, Capra V, Clunes MT, Rovati GE, Stankova J, Maj MC, Duffy DL (2016). «Гены пути цистеинил лейкотриенов, атопическая астма и реакция на лекарственные препараты: от популяционных изолятов до крупногеномных исследований ассоциации». Границы фармакологии. 7: 299. Дои:10.3389 / fphar.2016.00299. ЧВК 5131607. PMID 27990118.
дальнейшее чтение
- Намба Т., Нарумия С. (1993). «[Рецептор тромбоксана А2; структура, функция и распределение в тканях]». Ниппон Риншо. 51 (1): 233–40. PMID 8433523.
- Муругаппан С., Шанкар Х., Кунапули С.П. (2005). «Рецепторы тромбоцитов для адениновых нуклеотидов и тромбоксана А2». Семин. Тромб. Хемост. 30 (4): 411–8. Дои:10.1055 / с-2004-833476. PMID 15354262.
- Хирата М., Хаяси Ю., Ушикуби Ф. и др. (1991). «Клонирование и экспрессия кДНК рецептора тромбоксана А2 человека». Природа. 349 (6310): 617–20. Дои:10.1038 / 349617a0. PMID 1825698. S2CID 4368702.
- Райчоудхури М.К., Юкава М., Коллинз Л.Дж. и др. (1995). «Альтернативный сплайсинг дает расходящийся цитоплазматический хвост в человеческом эндотелиальном рецепторе тромбоксана А2». J. Biol. Chem. 270 (12): 7011. Дои:10.1074 / jbc.270.12.7011. PMID 7896853.
- Хирата Т., Какидзука А., Ушикуби Ф. и др. (1994). «Мутация Arg60 в Leu тромбоксанового рецептора A2 человека при доминантно наследуемом нарушении свертываемости крови». J. Clin. Вкладывать деньги. 94 (4): 1662–7. Дои:10.1172 / JCI117510. ЧВК 295328. PMID 7929844.
- Д'Анджело Д.Д., Дэвис М.Г., Али С., Дорн Г.В. (1994). «Клонирование и фармакологическая характеристика рецептора тромбоксана A2 из клеток K562 (хронический миелогенный лейкоз человека)». J. Pharmacol. Exp. Ther. 271 (2): 1034–41. PMID 7965765.
- Райчоудхури М.К., Юкава М., Коллинз Л.Дж. и др. (1994). «Альтернативный сплайсинг дает расходящийся цитоплазматический хвост в человеческом эндотелиальном рецепторе тромбоксана А2». J. Biol. Chem. 269 (30): 19256–61. PMID 8034687.
- Borg C, Lim CT, Yeomans DC, et al. (1994). «Очистка головного мозга крысы, аорты кролика и рецепторов тромбоксана А2 / простагландина Н2 тромбоцитов человека с помощью иммуноаффинной хроматографии с использованием антител против пептидов и рецепторов». J. Biol. Chem. 269 (8): 6109–16. PMID 8119956.
- Нусинг Р.М., Хирата М., Какидзука А. и др. (1993). «Характеристика и хромосомное картирование гена рецептора тромбоксана А2 человека». J. Biol. Chem. 268 (33): 25253–9. PMID 8227091.
- Funk CD, Furci L, Moran N, Fitzgerald GA (1994). «Точечная мутация в седьмом гидрофобном домене тромбоксанового рецептора А2 человека позволяет различать сайты связывания агонистов и антагонистов». Мол. Pharmacol. 44 (5): 934–9. PMID 8246916.
- Швенгель Д.А., Нури Н., Мейерс Д.А., Левитт Р.С. (1994). «Картирование сцепления человеческого тромбоксанового рецептора A2 (TBXA2R) с хромосомой 19p13.3 с использованием транскрибированных 3'-нетранслируемых полиморфизмов последовательности ДНК». Геномика. 18 (2): 212–5. Дои:10.1006 / geno.1993.1457. PMID 8288221.
- Offermanns S, Laugwitz KL, Spicher K, Schultz G (1994). «G-белки семейства G12 активируются через тромбоксан A2 и рецепторы тромбина в тромбоцитах человека». Proc. Natl. Акад. Sci. СОЕДИНЕННЫЕ ШТАТЫ АМЕРИКИ. 91 (2): 504–8. Дои:10.1073 / пнас.91.2.504. ЧВК 42977. PMID 8290554.
- Хирата Т., Ушикуби Ф., Какидзука А. и др. (1996). «Две изоформы рецептора тромбоксана A2 в тромбоцитах человека. Противоположное соединение с аденилатциклазой с различной чувствительностью к Arg60 и мутации Leu». J. Clin. Вкладывать деньги. 97 (4): 949–56. Дои:10.1172 / JCI118518. ЧВК 507140. PMID 8613548.
- Кинселла Б.Т., О'Махони ди-джей, Фицджеральд Г.А. (1997). «Альфа-изоформа рецептора тромбоксана А2 человека (ТР-альфа) функционально связывается с G-белками Gq и G11 in vivo и активируется изопростановым 8-эпи простагландином F2 альфа». J. Pharmacol. Exp. Ther. 281 (2): 957–64. PMID 9152406.
- Беккер К.П., Гарновская М, Геттис Т, Галушка П.В. (1999). «Связывание изоформ тромбоксанового рецептора A2 с Galpha13: влияние на связывание лиганда и передачу сигналов». Биохим. Биофиз. Acta. 1450 (3): 288–96. Дои:10.1016 / S0167-4889 (99) 00068-3. PMID 10395940.
- Барр С.Л., Вигг К.Г., Пакстис А.Дж. и др. (1999). «Сканирование генома для связи с синдромом Жиля де ла Туретта». Являюсь. J. Med. Genet. 88 (4): 437–45. Дои:10.1002 / (SICI) 1096-8628 (19990820) 88: 4 <437 :: AID-AJMG24> 3.0.CO; 2-E. PMID 10402514.
- Чжоу Х, Ян Ф, Тай ХХ (2001). «Фосфорилирование и десенсибилизация человеческого рецептора тромбоксана-альфа с помощью G-протеин-связанных рецепторных киназ». J. Pharmacol. Exp. Ther. 298 (3): 1243–51. PMID 11504827.
- Vezza R, Mezzasoma AM, Venditti G, Gresele P (2002). «Эндопероксиды простагландина и тромбоксан А2 активируют одни и те же изоформы рецепторов в тромбоцитах человека». Тромб. Haemost. 87 (1): 114–21. Дои:10.1055 / с-0037-1612953. PMID 11848439.
- Турек Дж. У., Халмос Т., Салливан Н. Л. и др. (2002). «Картирование сайта связывания лиганда для белка рецептора тромбоксана А2 человека». J. Biol. Chem. 277 (19): 16791–7. Дои:10.1074 / jbc.M105872200. PMID 11877412.
внешняя ссылка
- «Простаноидные рецепторы: ТП». База данных рецепторов и ионных каналов IUPHAR. Международный союз фундаментальной и клинической фармакологии.