Ингибиторы MTOR - MTOR inhibitors
| ингибиторы mTOR | |
|---|---|
| Класс препарата | |
 Шариковая модель сиролимуса, прототипа ингибитора mTOR | |
| Идентификаторы класса | |
| Использовать | Иммуносупрессия (рапамицин) |
| Механизм действия | mTOR торможение |
| Биологическая мишень | • FKBP12 • mTOR |
| В Викиданных | |
ингибиторы mTOR площадь класс препаратов которые препятствуют мишень рапамицина у млекопитающих (mTOR), который является серин / треонин-специфическая протеинкиназа что принадлежит семье фосфатидилинозитол-3 киназа (PI3K) родственные киназы (PIKKs). mTOR регулирует клеточный метаболизм, рост и пролиферацию путем формирования и передачи сигналов через два белковые комплексы, mTORC1 и mTORC2. Наиболее известные ингибиторы mTOR - это так называемые рапалоги (рапамицин и его аналоги), которые показали опухолевый ответ в клинических испытаниях против различных типов опухолей.[1]
История
Открытие mTOR было сделано несколько десятилетий назад во время исследования механизм действия своего ингибитор, рапамицин.[2][3] Рапамицин был впервые обнаружен в 1975 г. в образце почвы из г. Остров Пасхи из южной части Тихого океана, также известный как Рапа Нуи, откуда и произошло его название.[4] Рапамицин - это макролид, произведенный микроорганизм Streptomyces hygroscopicus и показал противогрибковый характеристики. Вскоре после открытия иммунодепрессивный были обнаружены свойства, которые позже привели к утверждению рапамицина в качестве иммунодепрессанта. В 1980-х годах было обнаружено, что рапамицин обладает противоопухолевой активностью, хотя точный механизм действия оставался неизвестным до многих лет.[2][5][6]
В 1990-х годах в этой области произошли драматические изменения из-за исследований механизма действия рапамицина и идентификации мишени для лекарственного средства.[4] Было обнаружено, что рапамицин ингибирует клеточная пролиферация и прогрессирование клеточного цикла. Исследования ингибирования mTOR являются развивающейся отраслью науки и имеют многообещающие результаты.[7]
Протеинкиназы и их ингибиторы
В целом, протеинкиназы делятся на две основные категории в зависимости от их субстратной специфичности, протеинтирозинкиназы и протеин серин / треонинкиназы. Киназы двойной специфичности являются подклассом тирозинкиназ.[8]
mTOR - это киназа из семейства киназы, относящиеся к фосфатидилинозитол-3-киназе (PIKK),[9] которые представляют собой семейство серин / треониновых протеинкиназ, с последовательностью, сходной с семейством липидкиназ, PI3K.[8] Эти киназы имеют разные биологические функции,[8] но все это большие белки с общей доменной структурой.[9]
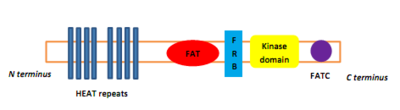
PIKK имеют четыре домена на уровне белка, что отличает их от других протеинкиназ. От N-конец к C-конец эти домены называются FRAP-ATM-TRAAP (FAT), киназный домен (KD), регуляторный домен PIKK (PRD) и FAT-C-концевой (FATC).[8] Домен FAT, состоящий из четырех α-спирали, является N-концевым по отношению к KD, но эта часть называется доменом связывания FKBP12-рапамицин (FRB), который связывает комплекс FKBP12-рапамицин.[8] Домен FAT состоит из повторов, называемых ВЫСОКАЯ ТЕМПЕРАТУРА (Хантингтин, Фактор удлинения 3, Подразделение протеинфосфатаза 2А и TOR1).[9] Специфические белковые активаторы регулируют киназы PIKK, но их связывание с киназным комплексом вызывает конформационное изменение, которое увеличивает доступ субстрата к домену киназы.[9]
Протеинкиназы стали популярными мишенями для лекарств.[10] Они были нацелены на открытие и разработку малая молекула ингибиторы и биопрепараты в качестве потенциальных терапевтических агентов. Низкомолекулярные ингибиторы протеинкиназ обычно предотвращают либо фосфорилирование белков субстраты или же аутофосфорилирование самой киназы.[11]
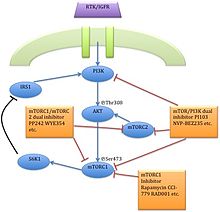
сигнальный путь mTOR
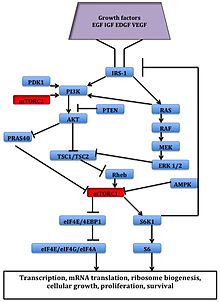
Оказалось, что факторы роста, аминокислоты, АТФ, и кислород уровни регулируют передачу сигналов mTOR. Несколько пути вниз по течению регулирующие развитие клеточного цикла,[12] перевод, инициация, ответы на транскрипционный стресс,[13] белок стабильность и выживаемость клеток сигнализируют через mTOR.

В серин / треонинкиназа mTOR является последующим эффектором PI3K / AKT пути и образует два разных мультипротеиновые комплексы, mTORC1 и mTORC2.[1] Эти два комплекса имеют отдельную сеть белковых партнеров, петли обратной связи, субстраты, и регуляторы.[15] mTORC1 состоит из mTOR и двух позитивных регуляторных субъединиц, раптор и LST8 млекопитающих (mlST8 ) и два негативных регулятора, богатые пролином AKT субстрат 40 (PRAS40) и DEPTOR.[1] mTORC2 состоит из mTOR, mLST8, mSin1, протор, риктор, и ДЕПТОР.[16]
mTORC1 чувствителен к рапамицину, но mTORC2 считается устойчивым и, как правило, нечувствительным к питательным веществам и энергетическим сигналам. mTORC2 активируется факторы роста, фосфорилаты PKCα, AKT и паксиллин, и регулирует активность малых GTPase, Rac, и Ро связанные с выживанием клеток, миграция и регулирование актиновый цитоскелет.
Сигнальный каскад mTORC1 активируется фосфорилированным AKT и приводит к фосфорилированию S6K1, и 4EBP1, что приводит к трансляция мРНК.[1]
Сигнальный путь mTOR при раке человека
Многие опухоли человека возникают из-за нарушения регуляции передачи сигналов mTOR и могут повышать чувствительность к ингибиторам mTOR.[17] Нарушение регуляции нескольких элементов пути mTOR, таких как PI3K усиление /мутация, PTEN Потеря функции, AKT сверхэкспрессия и S6K1, 4EBP1 и eIF4E сверхэкспрессия связана со многими типами рака. Поэтому mTOR - интересный терапевтическая цель для лечения множественного рака - как сами ингибиторы mTOR, так и в комбинации с ингибиторами других путей.[1]
Передача сигналов PI3K / AKT нарушается посредством различных механизмов, включая сверхэкспрессию или активацию рецепторы фактора роста, Такие как HER-2 (рецептор 2 эпидермального фактора роста человека) и IGFR (рецептор инсулиноподобного фактора роста), мутации в PI3K и мутации / амплификации AKT.[1] Фосфатаза-супрессор опухолей и гомолог тензина удалено на хромосома 10 (PTEN) является негативным регулятором передачи сигналов PI3K. При многих формах рака экспрессия PTEN снижена и может подавляться несколькими механизмами, включая мутации, потеря гетерозиготности, метилирование, и нестабильность белка.[16]
Ниже расположены эффекторы S6 киназы 1 (S6K1) mTOR, фактор инициации эукариот 4E-связывающий белок 1 (4EBP1) и фактор инициации эукариот 4E (eIF4E) связаны с клеточной трансформацией.[1] S6K1 является ключевым регулятором роста клеток, а также фосфорилирует другие важные мишени. Оба eIF4E и S6K1 включены в клеточная трансформация и их сверхэкспрессия была связана с плохим прогнозом рака.[16]
Разработка ингибиторов mTOR
С момента открытия mTOR было проведено много исследований по этому вопросу с использованием рапамицина и рапалогов для понимания его биологических функций.[15][18] Клинические результаты нацеливания на этот путь не были такими однозначными, как сначала казалось. Эти результаты изменили ход клинических исследований в этой области.[15]
Изначально рапамицин разрабатывался как противогрибковый препарат против грибковые микроорганизмы албиканс, Aspergillus fumigatus и Криптококк neoformans.[5] Через несколько лет были обнаружены его иммунодепрессивные свойства. Более поздние исследования привели к тому, что рапамицин стал основным иммунодепрессантом против отторжение трансплантата, вместе с циклоспорин А.[2] Использование рапамицина в сочетании с циклоспорином А усиливает профилактику отторжения у трансплантация почки. Следовательно, можно было использовать более низкие дозы циклоспорина, которые сводили к минимуму токсичность препарата.[5]
В 1980-х годах рапамицин был оценен терапевтическим отделением Национального института рака (NCI). Было обнаружено, что рапамицин обладает противоопухолевой активностью и является нецитотоксическим агентом с цитостатической активностью против нескольких типов рака человека.[5] Однако из-за неблагоприятных фармакокинетических свойств разработка ингибиторов mTOR для лечения рака в то время не была успешной.[3] С тех пор рапамицин также показал свою эффективность для предотвращения коронарной артерии. рестеноз и для лечения нейродегенеративные заболевания.[5]
Ингибиторы mTOR первого поколения
Разработка рапамицина как противоопухолевого средства снова началась в 1990-х годах с открытием темсиролимуса (CCI-779). Это было новое растворимое производное рапамицина, которое имело благоприятный токсикологический профиль у животных. Больше производных рапамицина с улучшенной фармакокинетикой и пониженным иммунодепрессивный с тех пор были разработаны эффекты для лечение рака.[5] Эти рапалоги включают темсиролимус (CCI-779), эверолимус (RAD001) и ридафоролимус (AP-23573), которые оцениваются при раке клинические испытания.[19] Аналоги рапамицина обладают такими же терапевтическими эффектами, как и рапамицин. Однако они улучшились гидрофильность и может использоваться для орального и внутривенное введение.[4] В 2012 Национальный институт рака перечислил более 200 клинических испытаний по проверке противоопухолевой активности рапалогов как монотерапия или как часть комбинированная терапия для многих типов рака.[7]
Рапалоги, являющиеся ингибиторами mTOR первого поколения, доказали свою эффективность в целом ряде доклинический модели. Однако успех в клинические испытания ограничивается лишь несколькими редкими формами рака.[20] Исследования на животных и клинические исследования показывают, что рапалоги в первую очередь цитостатический, и поэтому эффективны как стабилизаторы болезни, а не для регресса.[21] Частота ответа при солидных опухолях, когда рапалоги использовались в качестве монотерапии, была умеренной. Из-за частичного ингибирования mTOR, как упоминалось ранее, рапалоги недостаточны для достижения широкого и устойчивого противоракового эффекта, по крайней мере, при использовании в качестве монотерапия.[19][20][22]
Другая причина ограниченного успеха состоит в том, что в некоторых опухолевых клетках существует петля обратной связи между mTORC1 и AKT. Похоже, что ингибирование mTORC1 рапалогами не подавляет негативный отзыв цикл, который приводит к фосфорилирование и активация AKT.[18][23] Эти ограничения привели к разработке второго поколения ингибиторов mTOR.[7]
Рапамицин и рапалоги
Рапамицин и рапалоги (производные рапамицина) являются низкомолекулярные ингибиторы,[24] которые были оценены как противоопухолевые средства. Рапалоги имеют более благоприятный фармакокинетический профиль по сравнению с рапамицином, исходным препаратом,[3] несмотря на одинаковые сайты связывания для mTOR и FKBP12.[5]

Сиролимус
Бактериальный натуральный продукт рапамицин или сиролимус,[6] а цитостатический агент, использовался в комбинированной терапии с кортикостероиды и циклоспорин у пациентов, получивших трансплантация почки предотвращать отторжение органа как в США[25] и Европе,[26] из-за неудовлетворительных фармакокинетических свойств.[3] В 2003 г. Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США одобренные коронарные стенты с сиролимусом, которые используются у пациентов с сужением коронарные артерии, или так называемый атеросклероз.[27]
Недавно рапамицин показал свою эффективность в подавлении роста некоторых видов рака человека и линий мышиных клеток.[5] Рапамицин является основным ингибитором mTOR, но недавно разработанными аналогами рапамицина являются дефоролимус (AP23573), эверолимус (RAD001) и темсиролимус (CCI-779).[2]
Темсиролимус
Аналог рапамицина темсиролимус (CCI-779)[2] также является нецитотоксическим агентом, замедляющим пролиферацию опухоли.
Темсиролимус является пролекарством рапамицина. Он одобрен США. Управление по контролю за продуктами и лекарствами (FDA)[25] и Европейское агентство по лекарствам (EMA),[28] для лечения почечно-клеточного рака (ПКР). Темсиролимус более растворим в воде, чем рапамицин, поэтому его вводят внутривенно.[3][6] Он был одобрен FDA 30 мая 2007 г. для лечения запущенного ПКР.[6]
Темсиролимус также использовался в клинических испытаниях фазы I в сочетании с нератиниб, низкомолекулярный необратимый пан-HER ингибитор тирозинкиназы. В это исследование были включены пациенты, проходящие лечение от HER2 -усиленный рак молочной железы, немелкоклеточный рак легких с мутантным HER2 и другие распространенные солидные опухоли. Хотя общие токсичности включали тошнота, стоматит, и анемия; ответы были отмечены.[29]
Эверолимус

Эверолимус является вторым новым аналогом рапамицина.[2] С 30 марта 2009 г. по 5 мая 2011 г. FDA США одобрило эверолимус для лечения прогрессирующей почечно-клеточной карциномы после неудачного лечения сунитиниб или же сорафениб, субэпендимальная гигантоклеточная астроцитома (SEGA), связанный с туберозный склероз (TS) и прогрессирующие нейроэндокринные опухоли поджелудочной железы (PNET).[30] В июле и августе 2012 года были утверждены два новых показания: для поздних рецепторов гормонов, HER2-отрицательного рака молочной железы в сочетании с экземестаном, а также для педиатрических и взрослых пациентов с SEGA.[30] В 2009 и 2011 годах он также был одобрен во всем Европейском союзе для лечения распространенного рака груди, нейроэндокринных опухолей поджелудочной железы, прогрессирующей почечно-клеточной карциномы,[31] и SEGA у пациентов с туберозным склерозом.[32]
Ридафоролимус
Ридафоролимус (AP23573, MK-8669), или дефоролимус, является новейшим аналогом рапамицина и не пролекарство.[2] Как и темсиролимус, его можно вводить внутривенно, и предполагается, что пероральный состав предназначен для лечения саркома.[3] Его не было на рынке в июне 2012 года, поскольку FDA хотело проводить больше испытаний на людях из-за его эффективности и безопасности.[33]
Ингибиторы mTOR второго поколения
Второе поколение ингибиторов mTOR известно как АТФ-конкурентные ингибиторы киназы mTOR.[7] Двойные ингибиторы mTORC1 / mTORC2 предназначены для конкуренции с АТФ в каталитический сайт mTOR. Они ингибируют все киназозависимые функции mTORC1 и mTORC2 и, следовательно, блокируют активацию обратной связи передачи сигналов PI3K / AKT, в отличие от рапалогов, нацеленных только на mTORC1.[7][18] Эти типы ингибиторов были разработаны, и некоторые из них проходят клинические испытания. Как и рапалоги, они уменьшают белок перевод, ослабить клеточный цикл прогрессирование и тормозить ангиогенез во многих линиях раковых клеток, а также при раке человека. Фактически, они оказались более мощными, чем рапалоги.[7]
Теоретически наиболее важными преимуществами этих ингибиторов mTOR являются значительное снижение фосфорилирования AKT при блокаде mTORC2 и в дополнение к лучшему ингибированию mTORC1.[15] Однако есть и недостатки. Несмотря на то, что эти соединения были эффективны в клеточных линиях, нечувствительных к рапамицину, они показали лишь ограниченный успех в KRAS ведомые опухоли. Это говорит о том, что комбинационный Для лечения этих видов рака может потребоваться терапия. Еще один недостаток - это их потенциал. токсичность. Эти факты вызывают опасения по поводу долгосрочной эффективности этих типов ингибиторов.[7]
Тесное взаимодействие mTOR с путем PI3K также привело к разработке двойных ингибиторов mTOR / PI3K.[7] По сравнению с препаратами, которые ингибируют mTORC1 или PI3K, эти препараты обладают преимуществом ингибирования mTORC1, mTORC2 и всех каталитических изоформы из PI3K. Одновременное нацеливание на обе киназы снижает усиление регулирования PI3K, который обычно продуцируется при ингибировании mTORC1.[15] Было показано, что ингибирование пути PI3K / mTOR эффективно блокирует пролиферацию, индуцируя G1 арест в различных линиях опухолевых клеток. Сильная индукция апоптоз и аутофагия тоже был замечен. Несмотря на хорошие многообещающие результаты, существуют доклинические доказательства того, что некоторые виды рака могут быть нечувствительны к этому двойному ингибированию. Двойные ингибиторы PI3K / mTOR также могут иметь повышенную токсичность.[7]
Механизм действия
Исследования рапамицин так как иммунодепрессант позволили нам понять его механизм действия.[5] Подавляет Т-клетка распространение и пролиферативные ответы, вызванные несколькими цитокины, включая интерлейкин 1 (ИЛ-1), Ил-2, Ил-3, Ил-4, Ил-6, IGF, PDGF, и колониестимулирующие факторы (CSF).[5] Ингибиторы рапамицина и его аналоги могут прямо или косвенно воздействовать на рост опухоли. Прямое их воздействие на раковые клетки зависит от концентрации препарата и определенных клеточных характеристик. Непрямой способ основан на взаимодействии с процессами, необходимыми для опухоли. ангиогенез.[5]
Воздействие на раковые клетки
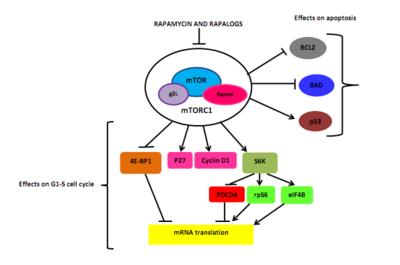
Рапамицин и рапалоги сшивают иммунофилин FK506 связывающий белок, такролимус или ФКБП-12, через его метокси группа. Комплекс рапамицин-FKBP12 вмешивается в домен FRB mTOR.[5][6] Молекулярное взаимодействие между FKBP12, mTOR и рапамицином может длиться около трех дней (72 часа). Ингибирование mTOR блокирует связывание вспомогательного белка raptor (регуляторно-связанный белок mTOR) с mTOR, но это необходимо для вниз по течению фосфорилирование S6K1 и 4EBP1.[5][22]
Как следствие, S6K1 дефосфорилирует, что снижает белок синтез и снижает смертность и размер клеток. Рапамицин также вызывает дефосфорилирование 4EBP1, что приводит к увеличению стр. 27 и уменьшение циклин D1 выражение. Это приводит к поздней блокировке G1 / S клеточный цикл. Было показано, что рапамицин вызывает гибель раковых клеток, стимулируя аутофагия или же апоптоз, но молекулярный механизм апоптоза в раковых клетках еще полностью не выяснен. Одно предположение о связи между ингибированием mTOR и апоптозом может быть связано с расположенной ниже мишенью S6K1, которая может фосфорилировать ПЛОХО, проапоптотическая молекула на Ser136.[5] Эта реакция разрушает связывание BAD с BCL-XL и BCL2, а митохондриальный ингибиторы смерти, приводящие к инактивации БАД[5] и снижение выживаемости клеток.[6] Также было показано, что рапамицин вызывает p53 -независимый апоптоз при некоторых типах рака.[5]
Влияние на ангиогенез опухоли
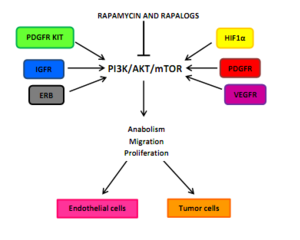
Ангиогенез опухоли зависит от взаимодействия между факторы роста эндотелиальных сосудов которые все могут активировать PI3K / AKT / mTOR в эндотелиальных клетках, перициты, или раковые клетки. Примером этих факторов роста являются ангиопоэтин 1 (ANG1), ANG 2, основной фактор роста фибробластов (bFGF), эфрин-B2, фактор роста эндотелия сосудов (VEGF), и члены фактор роста опухоли-β (TGFβ) надсемейство. Одним из основных стимулов ангиогенеза является гипоксия, приводящая к активации факторы транскрипции, индуцируемые гипоксией (HIF) и экспрессия ANG2, bFGF, PDGF, VEGF и VEGFR. Ингибирование трансляции HIF1α путем предотвращения PDGF / PDGFR и VEGF / VEGFR может быть результатом ингибирования mTOR. Блокада клеточного цикла G0-G1 может быть следствием инактивации mTOR в гипоксически-активированных перицитах и эндотелиальных клетках.[5]
Есть некоторые свидетельства того, что расширенная терапия рапамицином может влиять на AKT а также mTORC2.[2][34]
Влияние на химиотерапию
Фармакологическое подавление пути (mTOR) во время химиотерапии на мышиной модели предотвращает активацию примордиальных фолликулов, сохраняет функцию яичников и поддерживает нормальную фертильность с использованием клинически доступных ингибиторов INK и RAD. Таким образом, он помогает поддерживать фертильность во время химиотерапевтического лечения. Эти ингибиторы mTOR при введении в качестве предварительной обработки или совместного лечения со стандартной гонадотоксической химиотерапией помогают поддерживать фолликулы яичников в их изначальном состоянии.[35]
Структура отношения деятельности
Пипеколатная область структуры рапамицина, по-видимому, необходима для связывания рапамицина с FKBP12. Этот шаг необходим для дальнейшего связывания рапамицина с киназой mTOR, которая является ключевым ферментом во многих биологических действиях рапамицина.[36]
Высокое сродство связывания рапамицина с FKBP12 объясняется количеством водородные связи через два разных гидрофобный связывающие карманы, и это было выявлено с помощью рентгеновской кристаллической структуры соединения, связанного с белок. Структурные характеристики, общие для темсиролимуса и сиролимуса; то пипеколиновая кислота, трикарбонильная область от C13-C15, и лактон функциональные возможности играют ключевую роль в связывании групп с FKBP12.[19][37]
Самое важное водородные связи являются кислородом лактона карбонила в C-21 к основной цепи NH Иль56, амид карбонил у C-15 к фенольной группе на боковой цепи Tyr82, а гидроксильный протон на гемикетальный углерод, C-13, к боковой цепи Asp37.[37]

Структурные изменения в структуре рапамицина могут влиять на связывание с mTOR. Это может включать как прямое, так и непрямое связывание как часть связывания с FKBP12. Взаимодействие комплекса FKBP12-рапамицин с mTOR соответствует конформационной гибкости эффекторного домена рапамицина. Этот домен состоит из молекулярных областей, которые осуществляют гидрофобные взаимодействия с доменом FKB и Триен область от C-1-C-6, метоксигруппа у C-7 и метильные группы в C-33, C-27 и C-25. Все изменения макролидного кольца могут иметь непредсказуемые последствия для связывания и, следовательно, позволяют определять SAR по рапалогам проблематично.[37][38]
Рапамицин не содержит функциональных групп, которые ионизировать в pH диапазон 1-10 и поэтому нерастворимы в воде.[24] Несмотря на его эффективность в доклинических моделях рака, его плохая растворимость в воде, стабильность и длительный период полувыведения затрудняли его парентеральное использование, но разработка растворимых аналогов рапамицина преодолела различные препятствия.[2]
Тем не менее, аналоги рапамицина, одобренные для использования человеком, модифицированы по гидроксильной группе C-43 и показывают улучшение фармакокинетический параметры, а также свойства препарата, например, растворимость.[38]
Рапамицин и темсиролимус имеют схожие химические структуры и связываются с FKBP12, хотя их механизм действия отличается.[19]
Темсиролимус - дигидроксиметилпропионовый кислота сложный эфир рапамицина и его первого производного.[2] Следовательно, он более растворим в воде, и благодаря своей растворимости в воде его можно вводить внутривенно.[6][19]
Эверолимус имеет замещение гидроксиэтильной цепи O-2, а дефоролимус имеет оксид фосфина замена в положении C-43 в лактоновом кольце рапамицина.[19]
Дефоролимус (ридафоролимус) имеет C43 вторичный спиртовой фрагмент циклогексильной группы рапамицина, который был замещен фосфонатной и фосфинатной группами, предотвращая высокоаффинное связывание с mTOR и FKBP. Исследования компьютерного моделирования помогли синтезировать соединение.[6]
Неблагоприятные события
Лечение ингибиторами mTOR может осложняться нежелательными явлениями. Наиболее частыми нежелательными явлениями являются стоматит, сыпь, анемия, утомляемость, гипергликемия / гипертриглицеридемия, снижение аппетита, тошнота и диарея. Кроме того, интерстициальное заболевание легких является нежелательным явлением особой важности. mTORi-индуцированные ILD часто протекают бессимптомно (с аномалии матового стекла на КТ грудной клетки) или с легкими симптомами (с непродуктивным кашлем), но также может быть очень тяжелым. Описаны даже смертельные случаи. Следовательно, необходимы тщательная диагностика и лечение. Недавно был предложен новый подход к диагностике и лечению.[39]
Биомаркеры
Идентификация прогностической биомаркеры Эффективность лечения типов опухолей, чувствительных к ингибиторам mTOR, остается серьезной проблемой.[1][40]Возможные прогностические биомаркеры для ответ опухоли к ингибиторам mTOR, как описано в глиобластома, грудь и рак простаты клетки, может быть дифференциальной экспрессией белков пути mTOR, PTEN, AKT, и S6.[1] Таким образом, эти данные основаны на доклинических исследованиях, основанных на in vitro культивируемые линии опухолевых клеток, которые предполагают, что эффекты ингибиторов mTOR могут быть более выраженными при раковых заболеваниях, демонстрирующих потерю функций PTEN или PIK3CA мутации. Однако использование PTEN, PIK3CA мутации, а AKT-фосфо-статус для прогнозирования чувствительности к рапалогу не был полностью подтвержден в клинике. На сегодняшний день попытки определить биомаркеры ответа рапалога не увенчались успехом.[21]
Чувствительность
Клинические и трансляционные данные свидетельствуют о том, что чувствительные типы опухолей с адекватными параметрами и функциональными апоптоз пути, могут не потребоваться высокие дозы ингибиторов mTOR для запуска апоптоза. В большинстве случаев раковые клетки могут быть только частично чувствительны к ингибиторам mTOR из-за избыточности преобразование сигнала или отсутствие функциональных сигнальных путей апоптоза. В подобных ситуациях могут потребоваться высокие дозы ингибиторов mTOR. В недавнем исследовании пациентов с Карцинома почек, устойчивость к темсиролимусу была связана с низким уровнем p-AKT и p-S6K1, которые играют ключевую роль в активации mTOR. Эти данные убедительно свидетельствуют о количестве опухолей с активированным сигнальным путем PI3K / AKT / mTOR, которые не реагируют на ингибиторы mTOR. Для будущих исследований рекомендуется исключить пациентов с низким или отрицательным уровнем p-AKT из исследований с ингибиторами mTOR.
Текущих данных недостаточно для прогнозирования чувствительности опухолей к рапамицину. Однако имеющиеся данные позволяют охарактеризовать опухоли, которые могут не реагировать на рапалоги.[5]
АТФ-конкурентные ингибиторы киназы mTOR
Эти ингибиторы mTOR второго поколения связываются с АТФ-связывающим сайтом в киназном домене mTOR, необходимым для функций обоих mTORC1 и mTORC2, и в результате подавление сигнального пути mTOR. Благодаря способности PI3K и mTORC2 регулировать фосфорилирование AKT, эти два соединения играют ключевую роль в минимизации активации AKT с помощью обратной связи.[20]
Двойные ингибиторы mTOR / PI3K
Несколько так называемых двойных ингибиторов mTOR / PI3K (TPdI) были разработаны и находятся на ранней стадии. доклинические испытания и показать многообещающие результаты. Их развитию способствовали предыдущие исследования с селективными ингибиторами PI3K.[20] Активность этих малых молекул от активности рапалога отличается по способу блокирования как mTORC1-зависимого фосфолилирования S6K1, так и mTORC2-зависимого фосфорилирования остатка AKT Ser473.[1]
Двойные ингибиторы mTOR / PI3K включают: дактолизиб, BGT226, SF1126, PKI-587 и многие другие. Например, Новартис разработал соединение NVPBE235, которое, как сообщалось, ингибирует рост опухоли в различных доклинических моделях. Он усиливает противоопухолевую активность некоторых других препаратов, таких как винкристин.[20] Дактолизиб, по-видимому, эффективно ингибирует как дикую, так и мутантную форму PI3KCA, что предполагает его использование для лечения широких типов опухолей. Исследования показали превосходную антипролиферативную активность по сравнению с рапалогами и in vivo модели подтвердили эти мощные противоопухолевый эффекты двойных ингибиторов mTOR / PI3K.[1][7] Эти ингибиторы нацелены изоформы PI3K (p110α, β и γ) вместе с АТФ-связывающими сайтами mTORC1 и mTORC2 путем блокирования передачи сигналов PI3K / AKT, даже при типах рака с мутациями в этом пути.[7]
Двойные ингибиторы mTORC1 / mTORC2 (TORCdIs)
Новые mTOR-специфические ингибиторы появились в результате скрининга и открытие лекарств усилия. Эти соединения блокируют активность обоих комплексов mTOR и называются двойными ингибиторами mTORC1 / mTORC2.[20] Соединения с такими характеристиками, как сапанизертиб (кодовое название INK128), AZD8055 и AZD2014 вошли клинические испытания. Ряд этих ингибиторов киназы mTOR был изучен. Их структура происходит от морфолинопиразолопиримидинового каркаса.[20][22]Усовершенствования этого типа ингибиторов были сделаны путем замены морфолинов на мостиковые морфолины в пиразолопиримидиновых ингибиторах, и результаты показали повышение селективности к mTOR в 26000 раз.[22][41]
Ограничения ингибиторов mTOR нового поколения
Хотя новое поколение ингибиторов mTOR является многообещающим для противоопухолевой терапии и быстро проходит клинические испытания, существует множество важных факторов, определяющих их успех в клинике. Прежде всего, не существует предсказуемых биомаркеров в отношении этих ингибиторов. Похоже, что генетические детерминанты предрасполагают раковые клетки к чувствительности или устойчивости к этим соединениям. Опухоли, которые зависят от пути PI3K / mTOR, должны реагировать на эти агенты, но неясно, эффективны ли соединения при раке с отчетливыми генетическими повреждениями.[20]
Ингибирование mTOR - многообещающая стратегия лечения многих видов рака. Ограниченная клиническая активность селективных агентов mTORC1 делает маловероятным их влияние при лечении рака. Разработка конкурентных ингибиторов АТФ-катализа способна блокировать как mTORC1, так и mTORC2.[42]
Будущее
Ограничения доступных в настоящее время rapalogs привели к новым подходам к таргетингу mTOR. Исследования показывают, что ингибиторы mTOR могут обладать противораковой активностью при многих типах рака, таких как RCC, нейроэндокринные опухоли, рак молочной железы, гепатоцеллюлярная карцинома, саркома, и большая В-клеточная лимфома.[3]Одним из основных ограничений для разработки терапии ингибированием mTOR является то, что в настоящее время недоступны биомаркеры, позволяющие предсказать, какой пациент на них отреагирует. По-прежнему требуется лучшее понимание молекулярных механизмов, которые участвуют в ответе раковых клеток на ингибиторы mTOR, поэтому это возможно.[7]
Способом преодоления устойчивости и повышения эффективности нацеленных на mTOR агентов может быть стратификация пациентов и выбор комбинированной терапии. Это может привести к более эффективной и индивидуальной терапии рака.[1][7] Хотя необходимы дальнейшие исследования, нацеливание mTOR по-прежнему остается привлекательным и многообещающим терапевтическим вариантом для лечения рака.[7]
Смотрите также
- Мишень рапамицина у млекопитающих (mTOR)
- Путь PI3K / AKT / mTOR
- Сигнальный путь Akt / PKB
- Ингибитор PI3K
Рекомендации
- ^ а б c d е ж грамм час я j k л Популо, Елена; Лопес, Хосе Мануэль; Соарес, Паула (2012). «Сигнальный путь mTOR при раке человека». Международный журнал молекулярных наук. 13 (12): 1886–918. Дои:10.3390 / ijms13021886. ЧВК 3291999. PMID 22408430.
- ^ а б c d е ж грамм час я j Стримпакос, Алекс С .; Karapanagiotou, Eleni M .; Саиф М. Васиф; Сиригос, Костас Н. (2009). «Роль mTOR в лечении солидных опухолей: обзор». Отзывы о лечении рака. 35 (2): 148–59. Дои:10.1016 / j.ctrv.2008.09.006. PMID 19013721.
- ^ а б c d е ж грамм Юань, Жуйронг; Кей, Андреа; Берг, Уильям Дж; Лебволь, Дэвид (2009). «Нацеливание на онкогенез: разработка и использование ингибиторов mTOR в терапии рака». Журнал гематологии и онкологии. 2: 45. Дои:10.1186/1756-8722-2-45. ЧВК 2775749. PMID 19860903.
- ^ а б c Цанг, Чи Кван; Ци, Хайянь; Лю, Лерой Ф .; Чжэн, X.F. Стивен (2007). «Нацеливание на млекопитающих-мишень рапамицина (mTOR) для здоровья и болезней». Открытие наркотиков сегодня. 12 (3–4): 112–24. Дои:10.1016 / j.drudis.2006.12.008. PMID 17275731.
- ^ а б c d е ж грамм час я j k л м п о п q р Фэвр, Сандрин; Кремер, Гвидо; Раймонд, Эрик (2006). «Современные разработки ингибиторов mTOR как противоопухолевых средств». Обзоры природы Drug Discovery. 5 (8): 671–88. Дои:10.1038 / nrd2062. PMID 16883305.
- ^ а б c d е ж грамм час Vignot, S .; Faivre, S; Aguirre, D; Раймонд, Э (2005). «MTOR-таргетная терапия рака производными рапамицина». Анналы онкологии. 16 (4): 525–37. Дои:10.1093 / annonc / mdi113. PMID 15728109.
- ^ а б c d е ж грамм час я j k л м п Зайцева Екатерина Юрьевна; Валентино, Джозеф Д .; Гулхати, Пат; Эверс, Б. (2012). «Ингибиторы MTOR в терапии рака». Письма о раке. 319 (1): 1–7. Дои:10.1016 / j.canlet.2012.01.005. PMID 22261336.
- ^ а б c d е Лемпияйнен, Харри; Халазонетис, Танос Д (2009). «Новые общие темы в регулировании PIKK и PI3K». Журнал EMBO. 28 (20): 3067–73. Дои:10.1038 / emboj.2009.281. ЧВК 2752028. PMID 19779456.
- ^ а б c d Лавджой, Кортни А .; Кортес, Дэвид (2009). «Общие механизмы регуляции PIKK». Ремонт ДНК. 8 (9): 1004–8. Дои:10.1016 / j.dnarep.2009.04.006. ЧВК 2725225. PMID 19464237.
- ^ McConnell, J. L .; Вадзинский, Б. Э. (2009). «Ориентация на протеин-серин / треонинфосфатазы для разработки лекарств». Молекулярная фармакология. 75 (6): 1249–61. Дои:10,1124 / моль. 108,053140. ЧВК 2684880. PMID 19299564.
- ^ Грант, С. К. (2008). «Терапевтические ингибиторы протеинкиназ». Клеточные и молекулярные науки о жизни. 66 (7): 1163–77. Дои:10.1007 / s00018-008-8539-7. PMID 19011754.
- ^ Купер, Джеффри М. (2000). «Регуляторы развития клеточного цикла». Цитировать журнал требует
| журнал =(помощь) - ^ Юнгман, Матс (2007). «Ответ на стресс транскрипции». Клеточный цикл. 6 (18): 2252–7. Дои:10.4161 / cc.6.18.4751. PMID 17700065.
- ^ Lipton JO, Sahin M (октябрь 2014 г.). «Неврология mTOR». Нейрон. 84 (2): 275–291. Дои:10.1016 / j.neuron.2014.09.034. ЧВК 4223653. PMID 25374355.
Рисунок 1: Доменная структура киназы mTOR и компонентов mTORC1 и mTORC2
Рисунок 2: Путь передачи сигналов mTOR - ^ а б c d е Vilar, E .; Perez-Garcia, J .; Табернеро, Дж. (2011). «Расширяя границы пути mTOR: второе поколение ингибиторов». Молекулярная терапия рака. 10 (3): 395–403. Дои:10.1158 / 1535-7163.MCT-10-0905. ЧВК 3413411. PMID 21216931.
- ^ а б c Meric-Bernstam, F .; Гонсалес-Ангуло, А. М. (2009). «Ориентация на сигнальную сеть mTOR для лечения рака». Журнал клинической онкологии. 27 (13): 2278–87. Дои:10.1200 / Jco.2008.20.0766. ЧВК 2738634. PMID 19332717.
- ^ Хуанг, S; Houghton, PJ (2003). «Ориентация на передачу сигналов mTOR для лечения рака». Текущее мнение в фармакологии. 3 (4): 371–7. Дои:10.1016 / S1471-4892 (03) 00071-7. PMID 12901945.
- ^ а б c Баллоу, Лиза М .; Лин, Ричард З. (2008). «Ингибиторы рапамицина и mTOR-киназ». Журнал химической биологии. 1 (1–4): 27–36. Дои:10.1007 / s12154-008-0003-5. ЧВК 2698317. PMID 19568796.
- ^ а б c d е ж Брахманн, Саския; Фрич, Кристина; Майра, Савер-Мишель; Гарсия-Эчеверрия, Карлос (2009). «Ингибиторы PI3K и mTOR - новое поколение противоопухолевых средств таргетного действия». Текущее мнение в области клеточной биологии. 21 (2): 194–8. Дои:10.1016 / j.ceb.2008.12.011. PMID 19201591.
- ^ а б c d е ж грамм час Чжан, Ян-Цзе; Дуань, Янвэнь; Чжэн, X.F. Стивен (2011). «Нацеливание на домен киназы mTOR: второе поколение ингибиторов mTOR». Открытие наркотиков сегодня. 16 (7–8): 325–31. Дои:10.1016 / j.drudis.2011.02.008. ЧВК 3073023. PMID 21333749.
- ^ а б Wander, Seth A .; Хеннесси, Брайан Т .; Слингерленд, Джойс М. (2011). «Ингибиторы mTOR нового поколения в клинической онкологии: как сложность пути влияет на терапевтическую стратегию». Журнал клинических исследований. 121 (4): 1231–41. Дои:10.1172 / JCI44145. ЧВК 3069769. PMID 21490404.
- ^ а б c d Таннеру, Карунакар; Гурупрасад, Лалита (2011). «Генерация 3-D фармакофоров на основе лигандов и молекулярная стыковка ингибиторов киназы mTOR». Журнал молекулярного моделирования. 18 (4): 1611–24. Дои:10.1007 / s00894-011-1184-3. PMID 21805127.
- ^ Sutherlin, Daniel P .; Бао, Линда; Берри, Меган; Кастанедо, Жоржетта; Чукаури, Ирина; Дотсон, Дженна; Ребята, Адриан; Фридман, Лори; Голдсмит, Ричард; Гунзнер, Джанет; Хеффрон, Тимофей; Лесник, Джон; Льюис, Кристина; Матье, Симон; Мюррей, Джереми; Нономия, Джим; Панг, Джоди; Пегг, Ниль; Приор, Вэй Вэй; Руж, Лайонел; Салфати, Лоран; Сампатх, Дипак; Тянь, Цинпин; Цуй, Вики; Ван, Нан Чи; Ван, Шумей; Вэй, Бинькин; Визманн, Кристиан; Ву, Пинг; Чжу, Бин-Янь (2011). «Открытие сильнодействующего, селективного и перорально доступного фосфатидилинозитол-3-киназы класса I (PI3K) / млекопитающего-мишени ингибитора киназы рапамицина (mTOR) (GDC-0980) для лечения рака». Журнал медицинской химии. 54 (21): 7579–87. Дои:10.1021 / jm2009327. PMID 21981714.
- ^ а б Симамора, Пахала; Альварес, Джоан М; Ялковский, Сэмюэл Х (2001). «Солюбилизация рапамицина». Международный журнал фармацевтики. 213 (1–2): 25–9. Дои:10.1016 / s0378-5173 (00) 00617-7. PMID 11165091.
- ^ а б «Оранжевая книга: одобренные лекарственные препараты с оценками терапевтической эквивалентности». Управление по контролю за продуктами и лекарствами. Получено 25 сентября 2012.
- ^ «Рапамун». Европейское агентство по лекарствам. Получено 25 сентября 2012.
- ^ «Коронарный стент CYPHER, выделяющий сиролимус - P020026». Управление по контролю за продуктами и лекарствами. Получено 25 сентября 2012.
- ^ «Торисель». Европейское агентство по лекарствам. Получено 25 сентября 2012.
- ^ Ганди Л. и др. (2017). "MA04.02 Нератиниб ± Темсиролимус при HER2-мутантном раке легких: международное рандомизированное исследование фазы II". Журнал торакальной онкологии. 12 (1): S358-9. Дои:10.1016 / j.jtho.2016.11.398.
- ^ а б «Одобрение FDA для Эверолимуса». Национальный институт рака. 2009-04-21. Получено 20 сентября 2012.
- ^ "Афинитор". Европейское агентство по лекарствам. Получено 25 сентября 2012.
- ^ "Вотубия". Европейское агентство по лекарствам. Получено 25 сентября 2012.
- ^ «FDA хочет больше тестирования ридафоролимуса». Открытие и разработка лекарств. 2012-06-06. Получено 20 сентября 2012.
- ^ Гарсиа-Эчеверрия, Карлос (2011). «Блокирование пути mTOR: перспектива открытия лекарств». Сделки Биохимического Общества. 39 (2): 451–5. Дои:10.1042 / BST0390451. PMID 21428918.
- ^ Goldman, K. N .; Chenette, D .; Arju, R .; Duncan, F.E .; Keefe, D. L .; Grifo, J. A .; Шнайдер, Р. Дж. (2017). «Ингибирование mTORC1 / 2 сохраняет функцию яичников и фертильность во время генотоксической химиотерапии». Труды Национальной академии наук Соединенных Штатов Америки. 114 (12): 3186–3191. Дои:10.1073 / pnas.1617233114. ЧВК 5373380. PMID 28270607.
- ^ Ritacco, F. V .; Грациани, Э. И .; Саммерс, M. Y .; Забриски, Т. М .; Ю, К .; Бернан, В. С .; Картер, Г. Т .; Гринштейн, М. (2005). «Производство новых аналогов рапамицина с помощью биосинтеза, управляемого прекурсорами». Прикладная и экологическая микробиология. 71 (4): 1971–6. Дои:10.1128 / AEM.71.4.1971-1976.2005. ЧВК 1082568. PMID 15812028.
- ^ а б c Авраам, Роберт Т .; Гиббонс, Джеймс Дж .; Грациани, Эдмунд И. (2010). «Химия и фармакология рапамицина и его производных». В зале Майкл Н .; Таманой, Фуюхико (ред.). Структура, функция и регуляция комплексов TOR от дрожжей до млекопитающих. Ферменты. 27. С. 329–66. Дои:10.1016 / S1874-6047 (10) 27017-8. ISBN 978-0-12-381539-2.
- ^ а б Barrish, Joel C .; Картер, Перси; Ченг, Питер; и др., ред. (2010). Счета в открытии лекарств: тематические исследования в области медицинской химии. Кембридж: Королевское химическое общество. ISBN 978-1-84973-126-3.[страница нужна ]
- ^ Willemsen AE et al. Индуцированное ингибитором mTOR интерстициальное заболевание легких у онкологических больных: всесторонний обзор и практический алгоритм лечения. Международный журнал рака, 2015 г.
- ^ Дельбальдо, Екатерина; Альбер, Себастьян; Дрейер, Шанталь; Саблин, Мари-Поль; Серова Мария; Раймонд, Эрик; Faivre, Сандрин (2011). «Прогностические биомаркеры активности млекопитающих-мишеней ингибиторов рапамицина (mTOR)». Таргетированная онкология. 6 (2): 119–24. Дои:10.1007 / s11523-011-0177-6. PMID 21533544.
- ^ Новак, Павел; Коул, Дерек С .; Бройманс, Наташа; Бурсавич, Мэтью Г .; Курран, Кевин Дж .; Ellingboe, John W .; Гиббонс, Джеймс Дж .; Холландер, Ирвин; Ху, Юнбо; Каплан, Джошуа; Мальвиц, Дэвид Дж .; Торал-Барза, Лурдес; Verheijen, Jeroen C .; Заск, Арье; Чжан, Вэй-Го; Ю, Кер (2009). «Открытие сильнодействующих и селективных ингибиторов мишени рапамицина (mTOR) киназы млекопитающих». Журнал медицинской химии. 52 (22): 7081–9. Дои:10.1021 / jm9012642. PMID 19848404.
- ^ Альтман, Джессика К .; Сассано, Антонелла; Платаниас, Леонидас К. (14.06.2011). «Ориентация на mTOR для лечения AML. Новые агенты и новые направления». Oncotarget. 2 (6): 510–517. Дои:10.18632 / oncotarget.290. ЧВК 3248202. PMID 21680954.